We bestuderen mechanismen die de celfunctie reguleren, vooral met betrekking tot ziekten waarbij metabole stoornissen zijn betrokken, zoals type 2 diabetes en kanker. We bestuderen ook de metabole adaptatie bij dieren die weerstaan aan stress, zoals lage temperatuur, zuurstofgebrek en bevriezing, met medische implicaties voor orgaantransplantatie. Tot slot huisvesten we de centrale faciliteit voor massaspectrometrie op de Brusselse campus van de UCL, toegespitst op de analyse van proteinen.
Metformine is wereldwijd het meest voorgeschreven geneesmiddel voor de behandeling van type 2 diabetes, met als voornaamste effect de verlaging van de glucoseproductie door de lever. De werking van metformine kan gedeeltelijk worden verklaard door de activering van een enzym genaamd proteinekinase, in dit geval het AMP-geactiveerde proteïnekinase (AMPK). AMPK katalyseert de fosforylering van weer andere eiwitten, met name belangrijke enzymen van metabolische paden, die daardoor aan- of uitgeschakeld worden. In het algemeen schakelt de activering van AMPK de energie-producerende paden in weefsels en cellen aan, zoals de glycolyse en de vetzuuroxidatie, terwijl tegelijkertijd energie verbruikende processen, zoals de eiwitsynthese, worden uitgeschakeld. Zo herstelt AMPK de energiebalans binnen de cellen in reactie op een ATP depletie, zoals in het geval van anoxie of een intensieve spierinspanning. Dus haar belangrijkste rol is die van een sensor voor de homeostase van de cellulaire en de gehele lichaams-energie. Interessant is dat individuen die metformine nemen een lagere incidentie van bepaalde vormen van kanker vertonen, en er zijn steeds meer aanwijzingen dat AMPK activering metabolische dysfunctie bij bepaalde ziekte kan corrigeren. Daarom zijn farmaceutische bedrijven dan ook actief op zoek naar nieuwe geneesmiddelen die AMPK kunnen activeren voor de behandeling van type 2 diabetes en kanker.
Wij hebben belangrijke bijdragen geleverd aan het AMPK onderzoek door de identificatie van nieuwe targets in de controle van glucoseopname, glycogeensynthese, glycolyse en eiwitsynthese. De zoektocht naar AMPK targets is onze belangrijkste lijn van onderzoek, die we nastreven met behulp van state-of-the-art massaspectrometrische technieken. Met name in spier, lever en vetweefsel testen we ook nieuwe, directe AMPK activatoren, waaronder natuurlijke, plantafgeleide componenten, voor hun therapeutische werking bij de behandeling van diabetes en kanker. AMPK wordt met name geactiveerd tijdens spiercontractie en regelmatige lichaamsbeweging wordt daarom aanbevolen aan patiënten als behandeling voor type 2 diabetes.
Research topics:
Our group focuses on the control of cell function by reversible phosphorylation of proteins. Our main interests are the AMP-activated protein kinase (AMPK), insulin signaling via protein kinase B (PKB) and the role of fructose 2,6-bisphosphate (Fru-2,6-P2) in cell proliferation and cancer. We also investigate protein covalent modification and differential protein expression by electrospray ionization (ESI) mass spectrometry (MS) techniques.
1. AMPK: We are testing the effects of direct small-molecule AMPK activators and phytochemicals in muscle, liver and adipose tissue. We are also using direct AMPK activators in white adipose tissue to search for new targets and roles of AMPK.
2. Insulin signalling: We studied the role of protein kinase PKB in the stimulation of lipogenesis by insulin in white adipose tissue.
3. Role of Fru-2,6-P2 in the control of cell proliferation: We investigated the potential role of Fru-2,6-P2 in coupling glycolysis to cell proliferation in thymocytes stimulated by mitogens.
4. Mass spectrometry: We are developing mass spectrometry (MS)-based phosphoproteomics strategies. We routinely use MS for phosphorylation site identification and for the quantification of differential protein expression.

AMPK is a serine/threonine protein kinase involved in the control of cellular energy homeostasis. It stimulates ATP-producing pathways and inhibits energy-consuming processes (Fig.1). We contributed to this field by finding new AMPK targets. We demonstrated that the activation by AMPK of PFK-2, the enzyme responsible for Fru-2,6-P2 synthesis, participates in the stimulation of heart glycolysis by ischaemia (Pasteur Effect). We also showed that AMPK activation inhibits protein synthesis in anoxic rat hepatocytes and in ischaemic rat hearts. Protein synthesis inhibition in response to AMPK activation can partly be explained by increased eEF2 (eukaryotic elongation factor-2) phosphorylation leading to its inactivation, but this is not a direct effect (see below). Regulation of the upstream eEF2 kinase (eEF2K) is complex involving phosphorylation-induced activation/inactivation by kinases from various signalling pathways. We found that eEF2K, as well as being a direct AMPK substrate, is also controlled by AMPK-induced inhibition of mammalian target of rapamycin (mTOR) with implications in the biology of cancer.
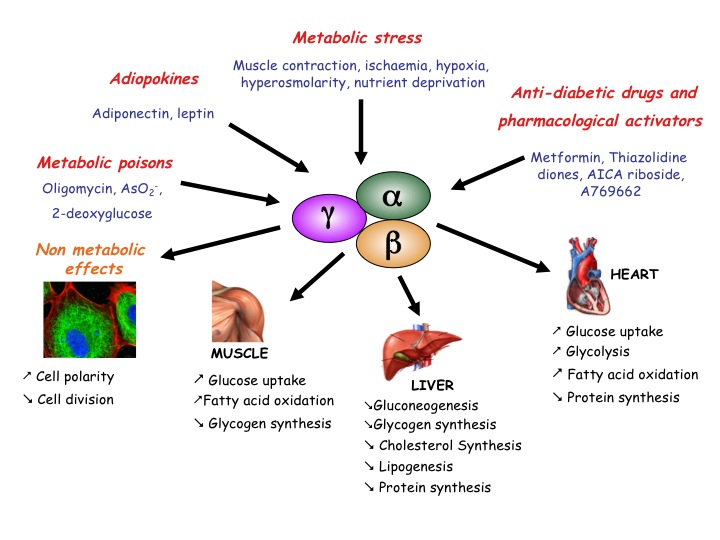
Figure 1: Conditions leading to AMPK activation in higher eukaryotes and some of its consequences.
We tested effects of direct AMPK activator compound 991 on hepatic glucagon signalling. Biguanides such as metformin had previously been shown to antagonize hepatic glucagon-stimulated cyclic AMP (cAMP) signalling independently of AMPK via direct inhibition of adenylate cyclase by AMP. We showed that incubation of hepatocytes with 991 decreased the glucagon-stimulated rise in cAMP, cyclic AMP-dependent protein kinase (PKA) activity and downstream PKA target phosphorylation. Moreover, incubation of hepatocytes with 991 increased the Vmax of cyclic nucleotide phosphodiesterase 4B (PDE4B) without affecting intracellular adenine nucleotide concentrations. The effects of 991 to decrease glucagon-stimulated cAMP concentrations and activate PDE4B were lost in hepatocytes deleted for both catalytic subunits of AMPK. Purified PDE4B was phosphorylated in vitro by AMPK at three sites, and by site-directed mutagenesis, Ser304 phosphorylation was shown to be important for activation. Our data suggest that AMPK activation in the liver could be beneficial by opposing short-term glucagon action via PDE activation to reduce cAMP as a new therapeutic strategy for the treatment of metabolic diseases associated with dysregulated cAMP/PKA signalling, such as type 2 diabetes (Fig.2). It is also noteworthy that cAMP signalling is important for the growth of certain cancers and that suppression of negative feedback mechanisms occurs during tumorigenesis.
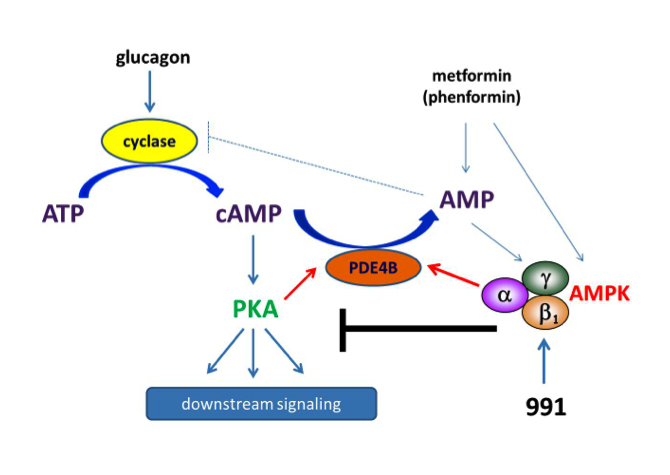
Figure 2: Schematic representation of the mechanisms by which biguanides and coumpound 991 antagonize glucagon signalling. Unlike biguanides, treatment with 991 activates AMPK without increasing cellular AMP levels. Both phenformin and 991 activate PDE in hepatocytes. Phosphorylation-induced activation of PDE4B by AMPK reduces glucagon-stimulated cAMP accumulation. As a consequence, PKA activation by glucagon and downstream signalling are decreased in hepatocytes incubated with 991, the effect being strictly AMPK-dependent.
We clarified the mechanisms by which AMPK activation leads to increased eEF2 phosphorylation to decrease protein synthesis by showing that increased eEF2 phosphorylation in MEFs was mainly due to direct activation of eEF2K and partly due to inhibition of mTOR signalling. Treatment of MEFs with AMPK activators led to eEF2K activation independently of AMPK probably via a rise in intracellular Ca2+. AMPK activated eEF2K by multi-site phosphorylation and newly identified Ser491/Ser492 was shown to be important for activation, leading to mTOR-independent inhibition of protein synthesis. Our study provides new insights into the control of eEF2K by AMPK, with implications for linking metabolic stress to decreased protein synthesis, a pathway that is of major importance in cancer cell survival (Fig.3).
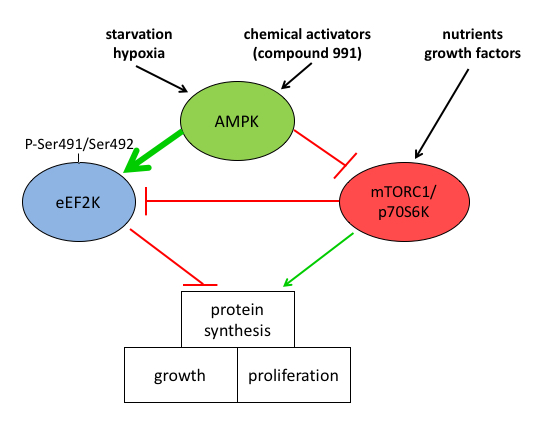
Figure 3: Mechanism of AMPK-induced inhibition of protein synthesis via eEF2K activation. AMPK activation inhibits protein synthesis needed for cell growth and proliferation. The mechanism involves both the indirect inhibition of mTOR and direct activation of eEF2K at the newly identified site(s) Ser491/Ser492.
We collaborated with the pharmaceutical company AstraZeneca (Mölndal, Sweden) to investigate whether the inhibition of AMP-metabolizing enzymes could be a strategy to achieve AMPK activation for the treatment of type 2 diabetes. In rat skeletal muscle incubated with small-molecule AMP-competitive AMP deaminase (AMPD) inhibitors and in muscles from mice bearing a whole body deletion of AMPD1, AMPK activation by electrical stimulation was potentiated, as was with the rise in AMP. However, no potentiation of AMP levels nor AMPK activation was seen in contracting muscles from whole body soluble 5'-nucleotidase cN-IA (NT5C1A) and cN-II (NT5C2) knockout mice. Overall, our findings indicate that inhibition of AMPD1 and NT5C enzymes would not be a viable strategy for achieving AMPK activation for the treatment of metabolic disorders.
For many years, we studied structure-function relationships and regulation by protein phosphorylation of the PFK-2/fructose-2,6-bisphosphatase (PFKFB) isoenzymes. We demonstrated the role of PKB in the mechanism by which insulin increases Fru-2,6-P2 content in heart via PFK-2 activation of the PFKFB2 isoenzyme. Using Akti-1/2 and MK-2206 PKB inhibitors, we also showed that PKB played an important role in the insulin-induced stimulation of glucose transport, glycogen synthesis and protein synthesis in incubated rat epitrochlearis muscle. In rat epididymal adipocytes, we studied the role of PKB in the stimulation of lipogenesis by insulin using Akti-1/2 and the MK-2206 inhibitor. Although incubation with the inhibitors blocked insulin-stimulated lipogenesis, the changes in phosphorylation states of some of the key enzymes were unaffected. Therefore, athough PKB activation is important, other pathways are involved in insulin-stimulated lipogenesis in white adipose tissue.
3. Role of Fru-2,6-P2 in the control of cell proliferation
Proliferating cells depend on glycolysis mainly to supply precursors for macromolecular synthesis. Mitogen stimulation of rat thymocytes with concanavalin A (ConA) led to time-dependent increases in lactate accumulation, Fru-2,6-P2 content, PFKFB3 and PFKFB4 protein levels and rates of cell proliferation and protein synthesis compared with resting cells. Although PFKFB3 could be phosphorylated at Ser461 by PKB in vitro leading to PFK-2 activation, PFKFB3 Ser461 phosphorylation was barely detectable in resting cells and only increased slightly in ConA-stimulated cells. On the other hand, PFKFB3 and PFKFB4 mRNA levels were decreased substantially by exposure of ConA-stimulated cells to low doses of MK-2206, suggesting control of expression of the two PFKFB isoenzymes by PKB. Treatment of ConA-stimulated thymocytes with PFK-2 inhibitor (3PO) or MK-2206 led to significant decreases in Fru-2,6-P2 content, medium lactate accumulation and rates of cell proliferation and protein synthesis. These data were confirmed by using siRNA knockdown of PFKFB3, PFKFB4 and PKB α/β in the more easily transfectable Jurkat E6-1 cell line. The findings suggest that increased PFKFB3 and PFKFB4 expression, but not increased PFKFB3 Ser461 phosphorylation, plays a role in increasing glycolysis in mitogen-stimulated thymocytes and implicate PKB in the upregulation of PFKFB3 and PFKFB4. The results support a role for Fru-2,6-P2 in coupling glycolysis to cell proliferation and protein synthesis in this model.
The development of mass spectrometry (MS) facilities within our laboratory, and for our Institute and University, has been an asset. Since the acquisition of an electrospray mass spectrometer in 1997, the application of mass spectrometry techniques has led to nearly 100 publications. In our own research, it enabled us to identify new phosphorylation sites in the AMPK complex and to demonstrate that in heart, insulin antagonized AMPK activation during ischaemia via PKB-induced phosphorylation of the AMPK catalytic α-subunits at Ser495/491. Our Institute has recently acquired the advanced high-resolution Orbitrap Fusion Lumos mass spectrometer from Thermo Scientific that will allow post-translational modification analysis and absolute quantification using isobaric tags.
We study differential protein expression by label-free multidimensional LC-MS. One application is the identification of physiological substrates of MsrP, a newly discovered methionine sulfoxide reductase. Twenty proteins that had one or more HOCl-oxidized Met residues that MsrP could reduce were identified, establishing that MsrP is able to repair a wide panel of structurally and functionally diverse periplasmic proteins.
We use phosphoproteomics to identify new targets downstream of signalling pathways. We developed a strategy of SDS protein extraction/trypsin digestion combined with strong cation exchange and hydrophilic interaction liquid chromatography (HILIC) followed by metal oxide affinity chromatography (MoAC) to enrich and concentrate phosphopeptides. The methodology was used to investigate changes in Brown adipose tissue (BAT) from thirteen-lined ground squirrels (Ictidomys tridecemlineatus), comparing euthermic squirrels with squirrels in deep torpor. Amongst the targets identified, pyruvate dehydrogenase (PDH) phosphorylation increased during ground squirrel hibernation, and this was confirmed by immunoblotting with phospho-specific antibodies. PDH phosphorylation leads to its inactivation, which suggests that BAT carbohydrate oxidation is inhibited during hibernation. Phosphorylation of hormone-sensitive lipase (HSL) also increased during hibernation, suggesting that HSL would be in a partly activated state in BAT to produce the fatty acids that are likely the primary fuel for thermogenesis during arousal.
Training opportunities:
Post-doc, Ph.D. and undergraduate students are welcome to contact Mark Rider about training and fellowship application opportunities. Note that some post-doctoral fellowships are available from the de Duve Institute post-doctoral program.
Selected links: Platform for protein analysis by mass spectrometry (MASSPROT)
Lai YC, Kviklyte S, Vertommen D, Lantier L, Foretz M, Viollet B, Hallén S, Rider MH.
Biochem J. 2014; 460(3):363-75.
Horman S, Vertommen D, Heath R, Neumann D, Mouton V, Woods A, Schlattner U, Wallimann T, Carling D, Hue L, Rider MH.
J Biol Chem. 2006; 281(9):5335-40.
Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, Hue L.
Biochem J. 2004; 381(Pt 3):561-79. Review.
Horman S, Browne G, Krause U, Patel J, Vertommen D, Bertrand L, Lavoinne A, Hue L, Proud C, Rider M.
Curr Biol. 2002; 12(16):1419-23.

 Download
Download
REGULATIE VAN DE CELFUNCTIE DOOR EIWITFOSFORYLATIE