La production du sang requiert la présence de petites protéines appelées cytokines. Les cytokines sont des molécules qui transmettent des signaux entre les cellules et induisent la croissance et la survie cellulaire. Elles agissent en se liant aux « récepteurs » des cellules cibles, qui fonctionnent comme des antennes qui transmettent un signal à l'intérieur de la cellule et finalement au noyau, pour l'expression des gènes cibles. Nous étudions comment ces récepteurs spécifiques s'agencent au niveau de la membrane et se lient à d'autres protéines intracellulaires, comme les Janus Kinases (JAKs), qui transmettent le signal au noyau. Nous avons constaté que des mutations au niveau de ces protéines, JAKs, ou des récepteurs eux-mêmes trompent les cellules et les induisent à se multiplier indéfiniment, ce qui donne naissance aux cancers du sang.
Introduction:
La voie de signalisation JAK-STAT:
Le génome humain code pour plus de 30 petites protéines appelées « cytokines », qui peuvent être circulantes ou agissent sur les cellules voisines pour réguler leur survie, leur croissance ou leur différenciation. Elles agissent en se liant aux récepteurs spécifiques qui se trouvent à la surface des cellules cibles. La réponse à une cytokine particulière dépendra de l'expression d'un récepteur spécifique.
Les cytokines induisent la dimérisation ou l'oligomérisation des récepteurs, qui rapprochent leurs domaines cytosoliques, lesquels sont pré-liés aux Janus Kinases. Ces dernières sont des «tyrosine kinases» qui sont liées à des séquences précises des récepteurs à l'intérieur de la cellule, mais cette interaction n'est pas covalente. Physiologiquement, les récepteurs forment des complexes avec les JAKs avant d'arriver à la surface cellulaire et sont inactifs en l'absence de cytokines, qui se lient normalement au domaine extracellulaire des récepteurs. Habituellement, les cellules exposent entre 500 et 2000 récepteurs d'un type de cytokines à leur surface. La cytokine, par exemple, l'érythropoïétine « Epo » (un facteur de croissance des précurseurs de globules rouges), se lie au domaine extracellulaire de l'EpoR (son récepteur spécifique), ce qui conduit à un changement de conformation du récepteur et le passage du dimère du récepteur de l’état inactif à l’état actif. Nous avons déterminé que ce changement conformationnel impliquait une rotation des récepteurs de telle sorte qu'une interface particulière des domaines transmembranaires soit formée. Suite à cela, les protéines JAK2 annexées au domaine cytosolique des récepteurs se rapprochent et peuvent se transphosphoryler sur leurs résidus tyrosine, ce qui déclenche alors l'activité enzymatique des JAKs (Figure 1). Ceci conduit à son tour, la phosphorylation des tyrosines du domaine cytosolique du récepteur. Celles-ci attirent des protéines de signalisation qui ont une affinité pour les tyrosines phosphorylées. Une fois liées aux récepteurs, ces protéines deviennent elles-mêmes phosphorylées, car elles sont assez proches des JAKs.
Une classe importante de protéines sont les « Signal Transducers and Activators of Transcription » : STATs, protéines transductrices de signal et activatrices de la transcription. Ces protéines, une fois phosphorylées par les JAKs et dimérisées migrent vers le noyau, où elles régulent l'expression des gènes. En bref, la voie JAK-STAT commence par la stimulation des récepteurs de cytokines, passe par l'activation des JAKs (il y a 4 Janus kinases codées dans notre génome) et se termine par l'activation des STATs (il y a 7 STATs codés dans notre génome). Selon le type de récepteur, différentes protéines JAKs et STATs sont activées, mais le modèle d’activation reste le même. En plus des STATs, plusieurs autres molécules adaptatrices et de signalisation telles que celles qui activent les voies Ras-MAP-kinase et la phosphatidylinositol-3'-kinase-Akt / protéine kinase B, sont attirées vers les JAKs et les récepteurs phosphorylés (figure 1). Le génome de la cellule, qui est intégré dans la chromatine reçoit un signal combiné, provenant des STATs activées ainsi que des autres voies. Ce signal exerce généralement un effet intense sur les gènes cibles, qui seront exprimés ; ceci se traduit au niveau cellulaire par la modification du taux de survie, de croissance et de différenciation des précurseurs des différentes lignées sanguines et des cellules immunitaires matures.
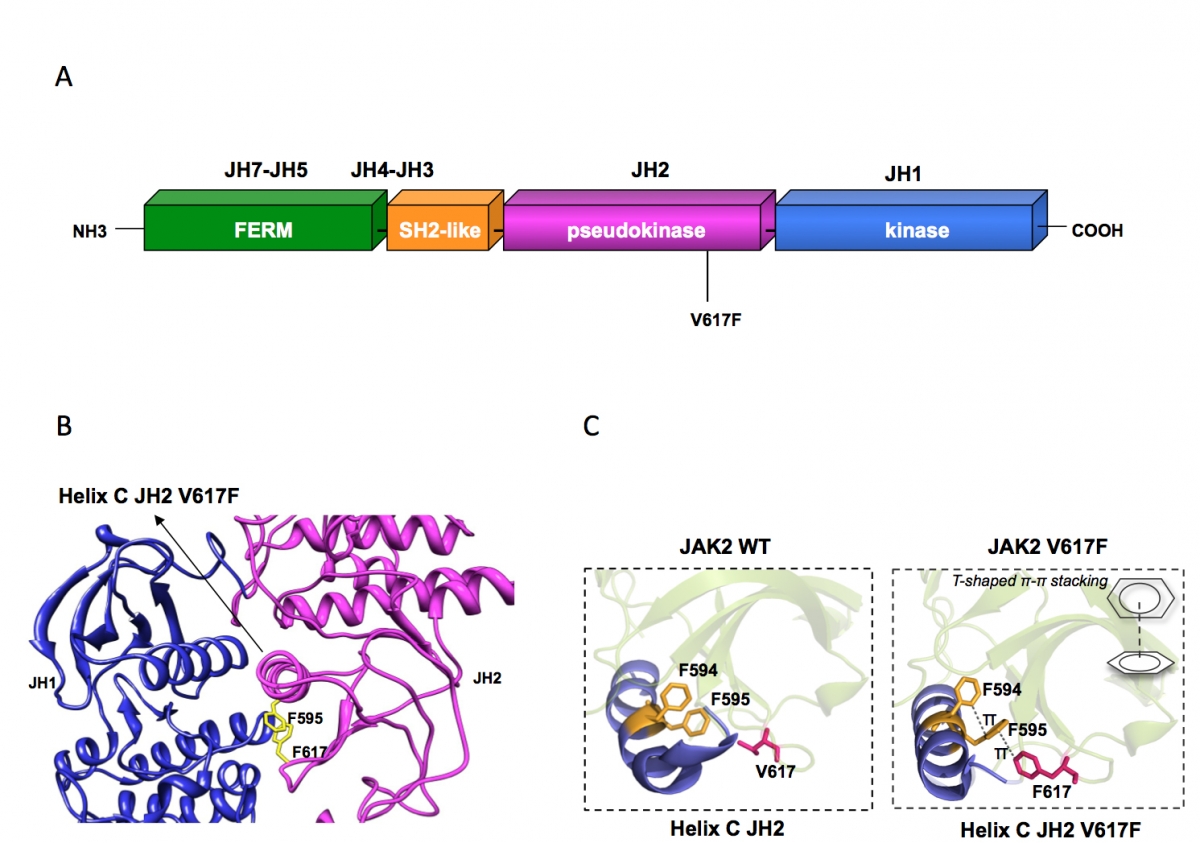
Figure 1:
(A) Janus kinase 2 contains several JAK homology domains, JH1, the kinase domain; JH2 the pseudokinase domain; JH3-JH4 the SH2-like domain and JH4-JH7, the FERM (band four point 1, ezrin, radixin, moesin)-like domain. The pesudokinase domain plays a major role in cytokine-dependent activation of the kinase domain, and was implicated in inhibiting the basal activity of the JH1 domain. The V617F mutation is activating the kinase activity of JH1, presumably by preventing the inhibition exerted by JH2 on JH1. The V617F mutation is detected in 98% of PV and approximately 50% of ET and PMF patients.
(B) The pseudokinase (JH2) and kinase (JH1) domains of JAK2 are modeled as adopting classical tyrosine kinase structures, interacting with each other and leading to JH1 inhibition. Residue F595 of the helix C of JH2 is required for constitutive activation of JAK2 V617F and of other mutated JAKs proteins, but not for cytokine activation of wild type JAK2. F595 plays a pivotal role in transmitting the conformational change in JH2 to JH1 (red arrow) and eventually in activating the kinase activity of JH1. The region around V617F and the middle of JH2 helix C surrounding F595 could become the target of inhibitors that might specifically decrease constitutive activation of JAK mutants.
(C) The X-ray crystal structures of JH2 and JH2 V617F solved by the Hubbard laboratory showed that in the wild type helix C is short and contains a kink while in JH2 V617F an aromatic stacking interaction occurs between F617 and helix C F595 and F594, which stabilizes and prolonges helix C. This conformational change associated with activation of JAK2 JH1 suggests that interruption of the aromatic stacking by small molecules would inhibit JAK2 V617F activity, but would not inhibit the function of wild type JAK2.
L'activation physiologique et pathologique des récepteurs de cytokines et de la voie de signalisation JAK-STAT:
Notre laboratoire, avec l’aide d'autres équipes, a décrit les interactions qui se produisent entre les protéines JAKs et les récepteurs, préalablement à leur activation, et qui sont importantes pour l'adressage du récepteur à la surface cellulaire. Nous avons montré que de nombreux récepteurs sont maintenus inactifs mais sont dimérisés (assemblés deux à deux) par des interactions entre les hélices transmembranaires. Suite à l'activation, les monomères du récepteur pivotent, conduisent à une rotation hélicoïdale rigide des régions cytosoliques juxtamembranaires et induisent donc l'activation des JAKs. Enfin, nous avons découvert des mutations au niveau des récepteurs (notamment le récepteur de l'érythropoïétine (EpoR) et le récepteur de la thrombopoïétine (TpoR)) qui provoquent la formation de dimères actifs et une signalisation constitutive. Cette activation est à l'origine d'un cancer. Certains de nos mutants TpoR W515 (W515A) sont en effet associés à des néoplasmes myéloprolifératifs humains graves (en anglais les « MPNs »). Cependant, nous avons constaté que la majorité des MPNs sont associés à l'unique mutation somatique activatrice de JAK2, c’est-à-dire « JAK2 V617F ». Ces résultats ont été également confirmés par d’autres équipes. Cette mutation est située dans le domaine pseudokinase de JAK2. Les protéines JAKs contiennent des domaines kinases à l'extrémité C-terminale, mais également des domaines pseudokinases qui précèdent les domaines kinases, ainsi que des régions qui sont nécessaires pour lier les JAKs aux récepteurs de cytokines (Figure 2). Le rôle des domaines pseudokinases reste inconnu, mais nous savons que ces domaines sont nécessaires pour le fonctionnement normal des JAKs dans la signalisation du récepteur aux cytokines. Nous avons également décrit des mutations activatrices de JAK1 (V658F) et TYK2 (V678F), qui se trouvent aux positions homologues à V617F dans JAK2.
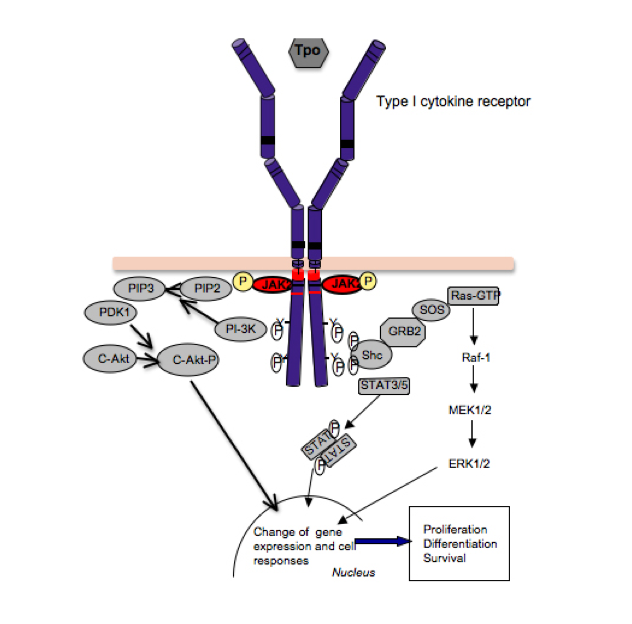
Figure 2:
Le récepteur de la thrombopoïétine (TpoR) contient un motif amphipathique unique (rouge), composé de la séquence RWQFP. En l'absence de Tpo, le motif se fixe à la membrane plasmique par l'intermédiaire de son résidu tryptophane (W515). En présence de Tpo, le récepteur pivote, et le motif se détache de la membrane, et W515 se retrouve à l'interface. La mutation de W515 en Leu, Ala, Lys et autres résidus, conduit à l'activation constitutive de TpoR. De même, la suppression du motif RWQFP (delta5 TpoR), conduit à l'activation du récepteur indépendamment du ligand. Les mutations de TpoR W515 sont les plus répandues dans les MPNs qui sont négatives pour la mutation JAK2 V617F. La mutation S505N est rare dans les MPNs, elle favorise la dimérisation des domaines transmembranaires de TpoR. La mutation T487A située sur le côté extracellulaire du domaine juxtamembranaire, est activatrice et a été détectée dans une lignée de cellules leucémiques.
Principaux défis et objectifs généraux:
Alors que les structures cristallines par diffraction aux rayons X existent pour de nombreux récepteurs de cytokines, des difficultés techniques rendent jusqu'à présent impossible, la visualisation à l'échelle atomique des structures des domaines transmembranaire et intracellulaire (cytosolique) des récepteurs de cytokines, ou des complexes entre les récepteurs et les JAKs. Néanmoins, le signal induit par les cytokines se transmet via les changements conformationnels des hélices transmembranaires, aux domaines cytosoliques et jusqu’aux JAKs.
Sans la compréhension exacte des interactions entre les résidus ainsi que les structures des complexes « récepteurs-JAKs », nous ne pouvons pas manipuler ces récepteurs à des fins médicales sinon simplement modifier les niveaux de cytokines disponibles. Mais selon les modèles, des effets partiels seraient bénéfiques, tandis que des actions complètes induites par les cytokines seraient préjudiciables.
Nos objectifs généraux sont les suivants: i) comprendre les interactions entre les domaines transmembranaires et juxtamembranaires et l’assemblage des récepteurs au niveau de la membrane; ii) comprendre comment les JAKs interagissent avec les récepteurs actifs et inactifs; iii) déterminer le rôle des domaines pseudokinases des JAKs dans la signalisation; iv) déterminer le mécanisme par lequel la mutation V617F du domaine pseudokinase de JAK2, active le domaine kinase, afin de trouver des inhibiteurs spécifiques de JAK2 V617F pour le traitement des MPNs; v) obtenir des données structurelles des complexes récepteurs de cytokines- JAKs et comprendre les mécanismes par lesquels des mutations dans TpoR W515K conduisent à l'activation du récepteur en l'absence du ligand.
Afin d'atteindre ces objectifs, nous suivons plusieurs approches. D'abord, nous avons créé un nouveau système qui nous permet de construire des dimères de récepteurs à partir d'orientations et d'interfaces connues. Nous accomplissons cela en remplaçant les domaines extracellulaires des récepteurs par des dimères enroulés en super-hélices (paires d'hélices) et nous créons des jonctions entre ces paires d'hélices (tenues fermement ensemble par des séquences répétées de forte affinité) et les domaines transmembranaires. Nous pouvons concevoir pour chaque dimère de récepteur, les sept interfaces dimèriques possibles et les tester biologiquement. Cela nous permet d'identifier les orientations dimériques actives, inactives et partiellement actives de plusieurs récepteurs de cytokines.
Deuxièmement, nous utilisons les techniques de mutagenèse dirigée et de pontage chimique afin de voir si les interfaces que nous prédisons sont effectivement retrouvées au niveau des segments membranaires ou dans les récepteurs entiers. Troisièmement, nous utilisons des tests biochimiques, des techniques de signalisation et des tests fonctionnels afin de mesurer l'activité des récepteurs et des mutants JAKs. Quatrièmement, nous utilisons des technologies de spectrométrie de masse (exemple : PhosphoScan et UbiScan) afin de déterminer le type de modification post-traductionnelle induite par les récepteurs de cytokines et par les JAKs dans différentes cellules. En cinquième lieu, nous utilisons des cellules primaires de patients et de souris afin de réaliser des essais de transplantation de moelle osseuse chez la souris (de type sauvage « WT », et génétiquement modifié). Ces tests nous permettront de valider nos résultats dans des systèmes in vivo et déterminer avec certitude les promoteurs qui lieraient les facteurs de transcription (tels que STAT5) et les gènes qui sont activés ou désactivés en aval.
Our major research directions are:
1. Orientation-dependent signaling of cytokine receptor: Ligand-imposed dimerization seems to be at the core of cytokine receptor activation. Using coiled coils we can impose predictable dimeric or tetrameric interfaces to the transmembrane and cytosolic domains of receptors and then test signaling and biologic effects of each conformation. Not only fully active conformations and their signaling outputs were identified, but also conformations that are partially active, that could be useful pharmacologically. We find that receptor sequences flanking the transmembrane domains function as switches for signaling activation and termination and aim to target them for pharmacologic modulation of receptor signaling.
2. Physiologic versus oncogenic signaling: Physiologic cytokine signaling is always transient. In contrast, mutations that permanently activate signaling of cytokine receptors are oncogenic. The reasons for the latter are not known. Using signaling assays, classical molecular biology and chromatin immunoprecipitation, we attempt to identify gene promoters that are targeted by the JAK-STAT pathway only in transformed cells.
3. JAK2 V617F and human myeloproliferative diseases: We and others have identified activating mutations in JAK2 (V617F) and in TpoR that are responsible for the majority of human myeloproliferative diseases such as Polycythemia Vera (PV), Essential Thrombocythemia (ET) and Primary Myelofibrosis (PMF). In these diseases, excessive numbers of red blood cells, platelets and granulocytes are formed, in the absence of cytokine stimulation. We try to understand how can one active mutant, JAK2 V617F, induce three related, but distinct, diseases and how do they evolve to acute leukemia. Furthermore, we aim to develop assays that can lead to identification of small molecules that only target JAK2 V617F for inhibition, and not wild type JAK2, which is crucial for blood formation.
Description of detailed projects
Blood formation and the functions of the immune system depend on cytokines and their specific receptors. These are transmembrane proteins that often form dimeric or oligomeric complexes and are coupled to one or several cytosolic tyrosine kinases belonging to the Janus kinase (JAK) family. The human genome codes for more than 30 cytokine receptors, four different JAKs and seven Signal Transducers and Activators of Transcription (STATs) proteins that shuttle to the nucleus and regulate gene expression.
Our broad interest is the understanding of the mechanisms that control assembly in the membrane of cell surface receptors that respond to extracellular cues. We study in detail the structure, function and orientation of several cytokine receptors, such as those for erythropoietin (Epo), thrombopoietin (Tpo), Granulocyte Colony Stimulating Factor (G-CSF), which function as homomeric complexes. We aim to identify: i) the structural basis for transmembrane signaling, especially how transmembrane and juxtamembrane sequences switch-on or -off cytokine receptor signaling; ii) what are the general rules by which hydrophobic transmembrane sequences interact in the membrane in a sequence-specific manner; and iii) the mechanisms of JAK attachment to receptors, and their subsequent activation, especially the role of pseudokinase domains in JAK kinase domain activation.
The laboratory identified constitutively active oncogenic mutants of JAK2 (V617F), JAK1 (V658F) and TYK2 (V658F) and of erythropoietin and thrombopoietin receptors, with some being involved in human blood cancers. Specifically the mechanisms by which JAK2 V617F and TpoR W515A/K/L mutants induce human Myeloproliferative Neoplasms (MPNs) and the role that constitutive STAT5 activation play in these diseases are actively pursued directions. Close interactions with clinicians and clinical biologists at St Luc Hospital allow the in-depth study of patient-derived cells.
The mechanisms of JAK2 V617F activation in human myeloproliferative neoplasms
Polycythemia Vera (PV), or the Vaquez-Osler disease, is characterized by excessive production of mature red cells and sometimes of platelets and granulocytes. Two other related diseases, Essential Thrombocythemia (ET) and Primary Myelofibrosis (PMF) are associated with excessive platelet, granulocyte production and fibrosis (scaring) of the marrow due to excessive myeloid cell proliferation, enzyme release and collagen secretion by marrow fibroblasts.
The unique acquired somatic JAK2 V617F mutation (Figure 1A) (1, 2) is responsible for >98% of Polycythemia Vera and for >50% of Essential Thrombocythemia (ET) and Primary Myelofibrosis (PMF) cases (2). The mutation in the pseudokinase domain activates the kinase domain and constitutive signaling (2, 3) in complexes between JAK2 V617F and cytokine receptors such as EpoR, TpoR and G-CSFR (Figure 2). The homologous V617F mutations in JAK1 and Tyk2 also enable these kinases to be activated without ligand-binding to cytokine receptors. JAK1 mutations have been described in adult acute T-lymphoblastic leukemia.
Figure 1:
(A) Janus kinase 2 contains several JAK homology domains, JH1, the kinase domain; JH2 the pseudokinase domain; JH3-JH4 the SH2-like domain and JH4-JH7, the FERM (band four point 1, ezrin, radixin, moesin)-like domain. The pesudokinase domain plays a major role in cytokine-dependent activation of the kinase domain, and was implicated in inhibiting the basal activity of the JH1 domain. The V617F mutation is activating the kinase activity of JH1, presumably by preventing the inhibition exerted by JH2 on JH1. The V617F mutation is detected in 98% of PV and approximately 50% of ET and PMF patients.
(B) The pseudokinase (JH2) and kinase (JH1) domains of JAK2 are modeled as adopting classical tyrosine kinase structures, interacting with each other and leading to JH1 inhibition. Residue F595 of the helix C of JH2 is required for constitutive activation of JAK2 V617F and of other mutated JAKs proteins, but not for cytokine activation of wild type JAK2. F595 plays a pivotal role in transmitting the conformational change in JH2 to JH1 (red arrow) and eventually in activating the kinase activity of JH1. The region around V617F and the middle of JH2 helix C surrounding F595 could become the target of inhibitors that might specifically decrease constitutive activation of JAK mutants.
(C) The X-ray crystal structures of JH2 and JH2 V617F solved by the Hubbard laboratory showed that the wild type helix C is short and contains a kink while in JH2 V617F an aromatic stacking interaction occurs between F617 and helix C F595 and F594, which stabilizes and prolonges helix C. This conformational change associated with activation of JAK2 JH1 suggests that interruption of the aromatic stacking by small molecules would inhibit JAK2 V617F activity, but would not inhibit the function of wild type JAK2.
Figure 2:
Signaling by cytokine receptors. The thrombopoeitin receptor signals as a homodimer that activates JAK2, which is bound to the cytosolic juxtamembrane domain. Activation of JAK2 leads to phosphorylation of tyrosine residues in the TpoR cytosolic domain, which recruit several signaling proteins like STAT5 and STAT3, which become substrates of JAK2, and upon tyrosine phosphorylation they dimerize, detach from the receptor and migrate into the nucleus, where they regulate gene expression. Cytokine receptors also activate, via JAK2, other pathways, such as the phosphatydylinositol-3’-kinase (PI-3’K)/akt/mTOR and ras-MAP-kinase. The integration of these signals in the nucleus, at the level of gene expression, induces survival, proliferation and differentiation of myeloid progenitors.
In addition to V617F we have described several other activating mutations on JAK2, V617I, L, M and W and modeled how such bulky residues could trigger the activation of JAK2 kinase domain (3). Interestingly, V617W is as strong as V617F, but requires a change in three base pairs to occur. In contrast, V617I is much weaker in inducing signaling than V617F/W. The V617I mutation can be obtained by one base pair change (3), has been detected in rare cases of myelofibrosis, and recently was identified in a family with hereditary thrombocytosis (4). JAK2 V617I promotes mainly STAT1 activation via TpoR, and very little EpoR activation, which may explain the limited thrombocytosis phenotype of this family (4). Primary cells from carriers of this heterozygous (germ line) mutation are being studied for signaling and function in the laboratory.
We aim to understand precisely how a pseudokinase domain mutation can induce kinase domain activation in a JAK. Besides the fundamental aspect, understanding of the mechanism of JAK activation via pseudokinase domain mutation would permit isolation of small molecule specific inhibitors of mutated JAK2 in myeloproliferative neoplasm patients, but not the wild type JAK2 (1), which is crucial for red blood cell and platelet formation. We identified pseudokinase residue F595 as absolutely required for constitutive activation by V617F, but not for cytokine-induced activation of JAK2/JAK2 V617F (5). A region around F617 and F595, involving the middle of helix C of the JAK2 pseudokinase domain might be a target for specific JAK2 V617F inhibition (Figure 1B and C). A recent X-ray crystal structure from the Hubbard and Silvennoinen laboratories has confirmed the major role of F595 in the conformation induced by V617F on helix C of JH2, with a complex three ring stacking interaction (F617-F595-F594) and prolongation by one turn of the helix C of JH2 (Figure 1C). Expression of segments of JAKs and cytokine receptors is pursued in insect and bacterial cells by D. Colau and M. Swinarska. We aim to obtain structural information of the receptor-JAK complexes and we take advantage of fusion proteins between coiled-coils and receptors' transmembrane domains and adopt different dimeric interfaces. These fusion proteins are active in functional studies and are more rigid than full-length receptors. They contain the full juxtamembrane and transmembrane sequences and they are membrane-anchored which bind and activate JAKs.
Pathologic TpoR signaling in myeloproliferative neoplasms
Thrombopoietin (Tpo) is a cytokine produced by the liver that is critical for regulation of the formation of platelet cells. Tpo also regulates the numbers of hematopoietic stem cells and other myeloid cells.
TpoR appears to be central to MPNs. First, observations of Jerry Spivak from Johns Hopkins University indicated that most MPN patients strongly down-modulate TpoR levels in megakaryocytes and platelets. Second, mutations in the TpoR intracellular juxtamembrane motif W515 were shown by our group to lead to constitutive activation of the receptor, and by several groups to induce severe in vivo MPN with myelofibrosis. Third, asparagine mutations, which induce dimerization of the transmembrane domain of TpoR also activate TpoR and one such mutation has been shown to be associated with familial ET. Fourth, alterations of TpoR traffic to the cell surface can induce thrombocytosis due to insufficient clearance of Tpo and high sensitivity of early megakaryocytes to high Tpo.
We have identified the mechanisms behind the down-modulation of TpoR in MPNs, and showed that JAK2 V617F induces ubiquitinylation, inhibition of recycling and degradation of TpoR (6). We discovered that Tpo can induce a strong antiproliferative effect in cells that express high JAK2 levels (6). This effect can be detected in postmitotic megakaryocytes (Plos Biol 2010, 8, e1000476). We showed that selection against the antiproliferative effect of Tpo occurs in JAK2 V617F cells, and is partially responsible for TpoR down-modulation in MPN cells, which then continue to proliferate in the presence of Tpo, unlike normal cells.
We employ a combination of Phospho-Scan (7) and Ubi-Scan approaches coupled to mass spectrometry in order to determine modifications in the profile of tyrosine phosphorylation and ubiquitinylation induced by Tpo ligand or expression of JAK2 V617F and TpoR W515 mutants.
A novel mechanism by which a tryptophan residue regulates TpoR activation
Several years ago we discovered that TpoR contained a unique motif at the junction between the transmembrane and cytosolic domains (RWQFP). By deletion or mutation of individual residues in this motif, Judith Staerk determined that this amphipathic insert actually prevented self-activation of the receptor and identified W515 in the motif as the key residue for this inhibitory function. (Blood 2006, 107(5), 1864-71). The question remained why would one W residue be so important in keeping a receptor inactive, and how could ligand addition defeat this inhibition. The question became even more relevant when several groups and ours detected TpoR W515L/K/A/R mutations in 5-8% of MPN patients that did not harbor JAK2 V617F. The answer came from two approaches. First, mutagenesis of W515 to all other residues showed the unique role played by this residue, in that even Y or F could not replace it (8). In W515L/A/K mutants, addition of W before or after the mutation could prevent activation. Second, biophysical experiments (analytical ultracentrifugation, solid-state NMR, infrared spectroscopy) performed by our collaborator, Prof. Steve O. Smith at SUNY Stony Brook, and fluorescence complementation studies performed in our laboratory by Vitalina Gryshkova showed that W515 actually regulates the orientation, tilt and dimerization of the upstream transmembrane helix, and prevents receptor activation (8). Taken apart, the transmembrane sequence of TpoR can dimerize, but this was not the case when the RWQFP insert was added or when W515 in the insert was mutated to K (8). Our data indicated that a W residue placed at the transmembrane-cytosolic junction could tilt the receptors so that the dimeric interface between transmembrane sequences can no longer be formed. Given that W residues were shown to promote transmembrane helix associations when present in the middle or on the extracellular side of membrane sequences, and given the many proteins that possess W residues at the cytosolic side of the transmembrane domain, we suggested a more general role for such W residues in preventing membrane domain self association. We pursue this direction by investigating the role of juxtamembrane W residues in several transmembrane proteins and we collaborate with Ahmed Essaghir in the group of Jean-Baptiste Demoulin at the de Duve Institute for bioinformatics of single and multi-span transmembrane proteins.
Mechanisms by which TpoR extracellular domain mutations induce severe hematological pathologies
TpoR contains in its extracellular domain two cytokine receptor modules, one distal that binds Tpo (D1-D2) and one proximal to the membrane (D3-D4), that appears to exert a negative role in receptor activation. Using domain deletions and swaps we showed that all 4 domains are required for physiologic traffic of the receptor to cell surface, and that deletion of D1-D2 significantly reduces traffic, while deletion of D3-D4 completely blocks traffic. Modeling and production of extracellular receptor segments indicated that the block in traffic was due to interactions with the membrane and the presence of transmembrane residues. The D1-D2 receptor resembles in its traffic a point mutant receptor that cannot be displayed at the surface (R102P) and that causes in humans congenital amegakaryocytic thrombocytopenia (CAMT) while others (P106L) which are expressed at lower than normal levels at the cell surface, but that paradoxically induce a thrombocytosis phenotype. We study the intracellular localization of these receptors in collaboration with Pierre Courtoy. We aim to understand why and where the intracellular traffic is blocked, as well as how and where these receptors signal, for example TpoR P106L, which seems to be resistant to Tpo-induced internalization. Preliminary evidence obtained recently, also in collaboration with William Vainchenker at Institut Gustave Roussy suggests that such receptors might take aberrant routes after cis-Golgi arrival and might be subject to a novel quality control step in the Golgi.
Determination of the interface and orientation of the activated dimeric cytokine receptors and downstream signaling pathways
While many X-ray crystal structures exist for G-protein coupled receptors and other membrane proteins with multiple transmembrane domains, no such structure could be obtained for single-span receptors. Extracellular domain structures exist for erythropoietin receptor (EpoR) or for G-CSFR, but it is not possible to relate those to transmembrane and cytosolic domains, and to their relative positioning in the inactive versus active states. To identify the residues that form the interface between the receptor monomers in an activated receptor dimer we have replaced the extracellular domain of the receptor (Figure 3A) with a coiled-coil dimer of a-helices (9 and Seubert et al. Mol Cell 2003). Because coiled-coils have a characteristic heptad repeat with hydrophobic residues at positions a (one), d (four), the register of the coiled-coil a-helices is imposed on the downstream TM a-helix and intracellular domain.
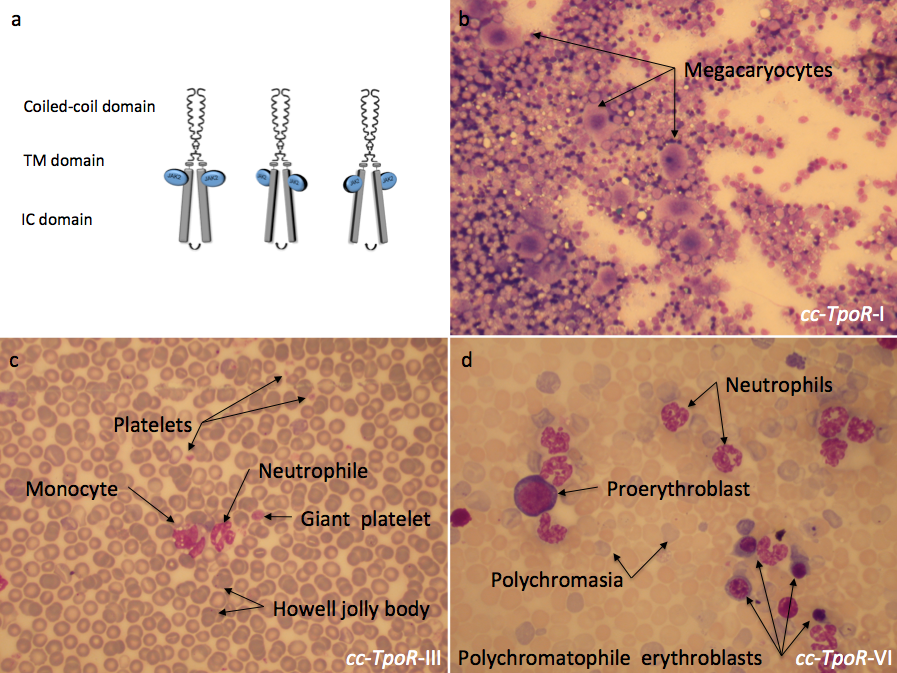
Figure 3:
Different orientations of the thrombopoietin receptor lead to distinct phenotypes in the bone marrow and in the peripheral blood. The fusion of the coiled coil protein to differently engineered transmembrane domains of the thrombopoietin receptor (TpoR) impose distinct dimeric orientations to the receptor (a). The effect induced by these chimeric receptors could be observed in the bone marrow and in the peripheral blood of mice reconstituted with bone marrow cells retrovirally transduced with the indicated cc-TpoR fusion proteins. cc-TpoR-I induced a strong megakaryocytic response with normal cellularity and a correct maturation of the myeloid lineages in the bone marrow (b). cc-TpoR-III exhibits granulocytosis, monocytosis and a very weak erythroblastosis in the peripheral blood (c). Although, the orientation adopted by cc-TpoR-VI is closed to the orientation of cc-TpoR-III, cc-TpoR-VI induced a stronger erythroblastosis and granulocytosis in the peripheral blood (d). a: May Grunwald stained bone marrow smear X 50; b and c: May Grunwald stained peripheral blood smear X 50. IC: intracellular, TM: transmembrane.
Each of the seven possible dimeric orientations will then be imposed by the coiled-coil on the fused TM and intracellular domain of receptors. We then express individually these dimers (seven for each receptor type) and test their function in cell lines, primary mouse and human cells and in vivo in mice. We expect that some dimer interfaces would be active and some inactive, reflecting requirements of the wild type receptor for activation. To prove that our predictions are correct, i.e. rotation imposed at the outset of the transmembrane domain is transmitted to the end of the transmembrane domain, we employed cysteine-mediated cross-linking and showed that indeed covalent dimers are formed via the cross-linker only when cysteine is in the predicted interface (9). This then allowed us to determine the active interface of the EpoR dimer, where only one interface was active.
Using this approach we have shown that TpoR can signal from several distinct dimeric interfaces, and that besides the normal dimeric interface (cc-TpoR-I), that leads to formation of platelets (Figure 3b), other interfaces promote signaling that leads to myeloproliferative and myelodysplastic disorders (Figure 3c and d) (10). One orientation (cc-TpoR-II) corresponds to the inactive receptor state. Interestingly, the dimer orientation that induces the highest levels of JAK2 activation, cc-TpoR-IV, also induces strong cell-to-cell adhesion and expansion of early hematopoietic progenitors. Our hypothesis is that in this dimeric orientation the receptor signals to maintain progenitors and possibly hematopoietic stem cells in the niche, and that signals induced by this dimer orientation might be recapitulating the quiescence-inducing effects of TpoR in HSCs. We will use this set of seven differently oriented TpoR dimers to dissect the signals induced by TpoR at the different stages of hematopoietic development and via the various downstream signaling proteins, JAK2, TYK2, STAT3, STAT5, MAP-kinase ERK1,2 and PI-3'-kinase/AKt/mTOR.
While all these pathways are activated by cytokines and mutant JAKs, we aimed to determine whether any of them could be essential for oncogenic proliferation. Within collaboration with Experimental Therapeutics Center in Singapore, we generated model cell lines that express JAK2 V617F or JAK2 with TpoR or EpoR, as well as cell lines that express TpoR W515L. These cells are screened for small molecule inhibitors at ETC. In collaboration with M. L. Choong, M. A. Lee and A. Matter at ETC we are showing that cells expressing JAK2 V617F are addicted to PI-3'-kinase signaling and that combinations of JAK2 inhibitors and pan type I PI-3'-kinase inhibitors are synergic in inhibiting proliferation of these transformed cells. Furthermore, the combinations are effective at inhibiting Epo-independent erythroid colony formation from JAK2 V617F knock-in mice or from MPN patient bone marrow cells.
Structure and function of juxtamembrane and transmembrane sequences of membrane proteins
We have previously shown that mutations in the extracellular juxtamembrane domain of EpoR can lead to constitutively active receptor mutants. Almost all cell surface EpoR, and a fraction of IL2/IL9/TpoR receptors exist on the cell surface as preformed ligand-independent complexes. Interactions between transmembrane domains are clearly maintaining EpoR dimers inactive. Introduction of asparagine residues in the transmembrane domain of TpoR defined two different interfaces that support TpoR activation and pointed to important differences between mouse and human TpoR, which seems to be more stringently controlled. Importantly, the human and not the mouse TpoR contains a His residue at the outset of the transmembrane domain, which is required for a small molecule agonist (Eltrombopag) to bind and activate the receptor. We study potential transmembrane interactions in the context of several transmembrane proteins, by cell surface immunofluorescence co-patching of differentially epitope tagged receptors along with protein ligation and protein fragment complementation. Preformed cytokine receptor oligomers might be important for supporting signaling by mutated JAKs in the absence of ligand.
In addition to cytokine receptors, we study the role of transmembrane dimerization in the amyloidogenic processing of Amyloid Precursor Protein (APP) in collaboration with the groups of Profs. Jean-Noël Octave in our university and Steven O. Smith (SUNY Stony Brook, NY). We identified three Gly-X-X-X-Gly motifs in the juxtamembrane and transmembrane domain of APP and showed that these consecutive motifs promote transmembrane helix dimerization and amyloidogenic processing of APP (J. Biol. Chem. 2008 283, 7733, Proc. Natl. Acad. Sci. USA 2009, 106, 1421).
Constitutive activation of JAK-STAT signaling pathways and genes targeted by STAT5 in transformed hematopoietic and patient-derived leukemia cells
Using chromatin immunoprecipitation and sequencing, Virginie Moucadel in the lab had shown that STAT5 contacts a substantially different set of promoters in cells that exhibit constitutive STAT5 activation, versus cells that respond to cytokines by acute STAT5 tyrosine phosphorylation (Moucadel and Constantinescu J Biol Chem 2005, 280(14):13364-73). We identified one specific target gene of constitutive active STAT5B signaling in megakaryocytes of MPN patients, namely Lipoma Preferred Partner (LPP) (10), a gene found to be translocated in rare leukemias. LPP is the host gene for miR-28, which we found to down-modulate TpoR translation, to inhibit translation of several proteins involved in megakaryocyte differentiation and to impair proplatelet formation (10). We found miR-28 to be pathologically overexpressed in 30% of MPNs (10). We identified the sites in LPP promoter that are required for induction by STAT5 and by CHIP on chip we identified several other genes that are regulated in the same way. Two of these genes appear to be associated with MPNs.
For induction of LPP/miR-28 transcription STAT5 and p53 must be synergistically bound to chromatin (10, 11). This occurs in transformed hematopoietic cells. Genome-wide association studies show that both STAT5 and p53 are co-localized on the chromatin at 463 genomic positions in proximal promoters. Binding of p53 to those promoters is dependent on STAT5 binding. We identified several novel STAT5-p53 target genes, namely LEP, ATP5J, GTF2A2, VEGFC, NPY1R and NPY5R, which appear to be pathologically expressed in platelets of MPN patients (11). We have concluded that persistently active STAT5 recruits normal and mutated p53 to novel promoters (Figure 4) leading to pathologic gene expression that differs from physiological STAT5 or p53 transcriptional programs.
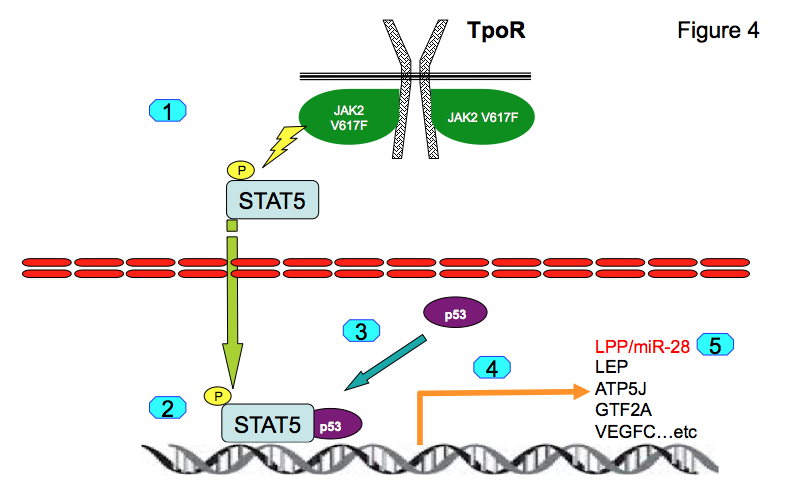
Figure 4:
For induction of LPP/miR-28 transcription STAT5 and p53 must be synergistically bound to chromatin (10, 11). This occurs in transformed hematopoietic cells. Genome-wide association studies show that both STAT5 and p53 are co-localized on the chromatin at 463 genomic positions in proximal promoters. Binding of p53 to those promoters is dependent on STAT5 binding. We identified several novel STAT5-p53 target genes, namely LEP, ATP5J, GTF2A2, VEGFC, NPY1R and NPY5R, which appear to be pathologically expressed in platelets of MPN patients (11). We have concluded that persistently active STAT5 recruits normal and mutated p53 to novel promoters (Figure 5) leading to pathologic gene expression that differs from physiological STAT5 or p53 transcriptional programs.
Interaction with St Luc Hospital clinicians and clinical biologists:
Identification of the molecular bases of MPNs without known molecular cause
At present, our laboratory is performing under the auspices of an ARC grant (Action de Recherche Concertée of the Université catholique de Louvain) with the St Luc Hospital departments of Hematology (Prof. Cédric Hermans, Prof. Augustin Ferrant, Dr. Laurent Knoops), Clinical Biology (Prof. Dominique Latinne, Dr Hélène Antoine-Poirel) and groups of de Duve Institute (Prof. Mark Rider, Prof. Jean-Baptiste Demoulin) a large study on the presence and signaling of JAK2, TpoR, and growth factor receptor mutations in patients with myeloproliferative neoplasms. Next generation sequencing will be employed for well-investigated patients, using primary cells that are characterized for functional defects and that do not harbor known mutations in order to unravel novel molecular defects in MPNs and leukemias.
References
Staerk J, Defour JP, Pecquet C, Leroy E, Antoine-Poirel H, Brett I, Itaya M, Smith SO, Vainchenker W, Constantinescu SN.
EMBO J. 2011; 30(21):4398-413.
Dusa A, Mouton C, Pecquet C, Herman M, Constantinescu SN.
PLoS One. 2010; 5(6):e11157.
Pecquet C, Staerk J, Chaligné R, Goss V, Lee KA, Zhang X, Rush J, Van Hees J, Poirel HA, Scheiff JM, Vainchenker W, Giraudier S, Polakiewicz RD, Constantinescu SN.
Blood. 2010; 115(5):1037-48.
Staerk J, Lacout C, Sato T, Smith SO, Vainchenker W, Constantinescu SN.
Blood. 2006; 107(5):1864-71.
James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, Garçon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W.
Nature. 2005; 434(7037):1144-8.
Seubert N, Royer Y, Staerk J, Kubatzky KF, Moucadel V, Krishnakumar S, Smith SO, Constantinescu SN.
Mol Cell. 2003; 12(5):1239-50.

 Telecharger
Telecharger
TRANSDUCTION DU SIGNAL ET HEMATOLOGIE MOLECULAIRE
