Ons onderzoek is gericht op tumorimmunologie, in het bijzonder op twee belangrijke aandachtspunten bij de ontwikkeling van immuuntherapie bij kanker: (i) wat zijn de mechanismen bij de productie van tumorantigenen en (ii) hoe komt het dat tumoren vaak toch in staat zijn een immunitaire aanval af te weren en hoe kunnen wij dit voorkomen?
Ons immuunsysteem beschermt ons tegen allerlei ziektes. Een belangrijke eigenschap van het systeem is dat indringers, zoals bacteriën of virussen, worden herkend als lichaamsvreemd. Ons immuunsysteem herkent ook kankercellen als vreemde lichamen die vernietigd moeten worden. Uit het werk dat onze teams verricht hebben in de jaren 1990 is duidelijk gebleken dat kankercellen beschikken over “merkers”, ook tumorantigenen genoemd, die de kankercellen onderscheiden van de normale cellen van het organisme. Dit heeft de weg geopend voor nieuwe therapeutische benaderingen, gericht op het versterken van de immuunrespons tegen kankers. Sommige van deze benaderingen maken vandaag deel uit van de gangbare klinische praktijk en maken het mogelijk om patiënten met een vergevorderd metastatisch melanoom te redden.
Ondanks de klinische successen wordt immuuntherapie op dit moment echter nog beperkt door twee fenomenen. Het eerste fenomeen is de inductie van toxische verschijnselen van auto-immunitaire aard. Bij bepaalde vormen van immuuntherapie wordt naast de immuniteit tegen tumoren ook de auto-immuniteit gestimuleerd, wat betekent dat gezonde weefsels aangevallen worden door het immuunsysteem. De tweede beperking komt doordat heel wat kankers een weerstand ontwikkelen tegen de immuunrespons.
Ons huidig onderzoek richt zich op het doorgronden en omzeilen van deze twee obstakels. Voor het eerste obstakel bestuderen we volgens welke mechanismen de tumorantigenen, de “merkers” van de tumorcel, worden geproduceerd. Dit onderzoek maakt het mogelijk om te definiëren welke de meest specifieke antigenen van de tumoren zijn en welke we best tot doel nemen om een immuunrespons tegen kanker te stimuleren die vrij is van auto-immune toxiciteit. Aangaande het tweede obstakel bestuderen we in detail de micro-omgeving van de tumoren. Haar immuunsuppressieve aard lijkt de doeltreffendheid van de immuuntherapie duidelijk te beperken. We ontwikkelen verschillende therapeutische strategieën om deze immuunsuppressie te bestrijden.
De tumorantigenen worden voornamelijk herkend door gespecialiseerde witte bloedcellen, de cytotoxische T-lymfocyten (CTL). Deze witte bloedcellen detecteren peptiden (kleine stukjes proteïnen) op de oppervlakte van de kankercellen. Deze peptiden bestaan uit acht tot tien aminozuren. Ze worden op het celoppervlak gepresenteerd door klasse I moleculen van het “major histocompatibility complex” (MHC, ook wel HLA genoemd bij de mens, een groep proteinen die een belangrijke rol spelen in het herkennen van compatibele en niet-compatibele elementen in het lichaam).
De peptiden zijn meestal afkomstig van de afbraak van de intracellulaire proteïnen door proteasomen, eiwitcomplexen die zich in het cytoplasma en in de kern bevinden. Vervolgens worden de peptiden getransporteerd naar het celoppervlak, na koppeling aan de MHC-moleculen. Ons onderzoek heeft aangetoond dat er verschillende vormen van het proteasoom bestaan en dat deze verschillen in hun capaciteit om peptiden te produceren die overeenstemmen met de tumorantigenen. Daarom hangen de antigenen die zich aan de oppervlakte van de kankercellen bevinden deels af van de proteasoomsamenstelling van deze cellen. We bestuderen deze fenomenen in detail om beter te kunnen bepalen welke antigenen we als doel moeten nemen voor de immuuntherapie. We bestuderen ook een nieuwe functie van het proteasoom die we hebben ontdekt, namelijk de productie van peptiden uit niet-contigue fragmenten in de moederproteïne, volgens het principe van “cut and paste”.
Verder bestuderen we ook hoe bepaalde kankers proberen te ontsnappen aan de immuunrespons door “zich te verstoppen”. Deze kankers verbergen immers de expressie van de tumorantigenen die hen “verraden" aan het immuunsysteem. Hierdoor ontmaskeren ze andere antigenen die wij nu proberen te typeren om de kankers beter te kunnen bestrijden. Het lijkt wel of we een soort van schaakspel tegen de kanker spelen.
Het is inmiddels duidelijk dat wanneer immuuntherapie niet werkt bij bepaalde kankerpatiënten, dit vaak te wijten is aan immuunsuppressieve mechanismen in de tumor zelf, die een goede werking van het immuunsysteem verhinderen. Wij bestuderen deze mechanismen in muismodellen, en dan voornamelijk in een transgeen model van een induceerbaar melanoom dat wij ontwikkeld hebben. Hieruit hebben we geleerd dat de tumoren de lymfocyten die hen komen vernietigen op een selectieve manier doden door apoptose.
We bestuderen de moleculaire mechanismen die verantwoordelijk zijn voor deze fenomenen van immuunsuppressie. Zo konden we al aantonen dat een van deze mechanismen zich baseert op de afbraak van een aminozuur dat de lymfocyten absoluut nodig hebben, namelijk tryptofaan. De tumoren brengen het enzym indoleaminedioxigenase (IDO) tot uitdrukking dat tryptofaan snel afbreekt en zo het immuunsysteem verlamt. In een spin-off die we in 2012 hebben opgericht, iTeos Therapeutics, onderzoeken we de inhibitoren van IDO die gebruikt zouden kunnen worden in kankertherapie. We proberen ook de mechanismen te begrijpen die verantwoordelijk zijn voor de uitdrukking van dit enzym door de kankers.
We hebben ook andere mechanismen van immuunsuppressie ontdekt waarvan we de moleculaire actoren bestuderen om ze te kunnen bestrijden.
In recent years, cancer immunotherapy made tremendous progresses and reached the clinical arena by showing its ability to prolong the survival of advanced cancer patients. This was largely based on the discovery of tumor antigens by our groups in Brussels in the nineties, a finding that demonstrated that our immune system has the capacity to recognize cancer cells as foreign bodies. These recent clinical results have generated enormous interest in the oncology field and the pharmacological industry. Yet, clinical benefits remain limited to a subset of patients, and further research is needed to understand the reason for this. Our current research focuses on two aspects that are relevant to this question. The first is the processing of tumor antigens, i.e. the intracellular mechanisms responsible for the expression of antigens at the surface of tumor cells. The second is the tumor microenvironment, whose immunosuppressive properties emerge as a major reason why many patients currently do not benefit from cancer immunotherapy.
I. PROCESSING OF TUMOR ANTIGENS (Group Nathalie Vigneron)
1. Peptide splicing by the proteasome
N. Vigneron, V. Stroobant, A. Michaux
Tumor antigens relevant for cancer immunotherapy consist of peptides presented by MHC class I molecules and derived from intracellular tumor proteins. They result from degradation of these proteins, mainly exerted by the proteasome. We have identified a new mode of production of antigenic peptides, which involves the splicing of peptide fragments by the proteasome (1). Peptide splicing occurs in the proteasome catalytic chamber through a reaction of transpeptidation involving an acyl-enzyme intermediate (Figure 1). Splicing of peptide fragments can occur in the forward or reverse order to that in which they appear in the parental protein (2). We have now described five spliced peptides, three of which are spliced in the reverse order (3, 4) One of these peptides also contains two additional post-translational modifications, resulting in the conversion of asparagines into aspartic acids, through a process a N-glycosylation/deglycosylation (3). We also showed that the splicing reaction required a minimal size of 3 amino acids for the fragments to splice (4). Both the standard proteasome and the immunoproteasome have the ability to splice peptides. However, their ability to produce a given spliced peptide varies according to their ability to perform the relevant cleavages to liberate the fragments to splice.
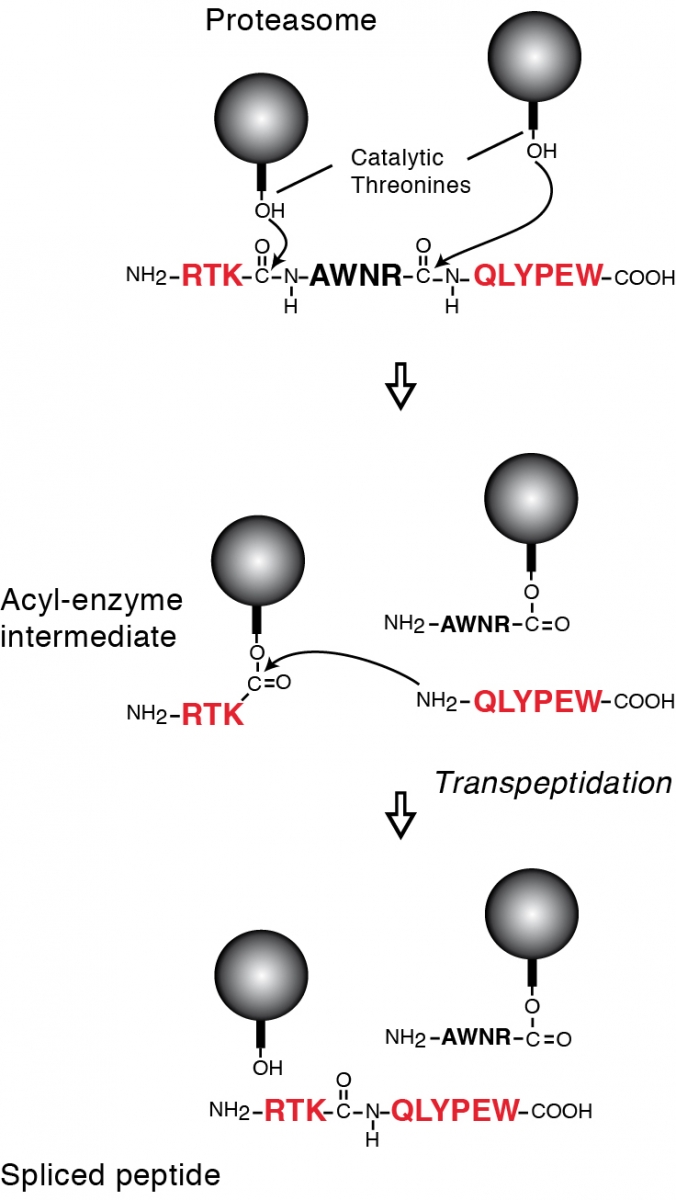
Legend: Model of the peptide-splicing reaction in the proteasome. The active site of the catalytic subunits of the proteasome is made up of the side-chain of a threonine residue, which initiates proteolysis by performing a nucleophilic attack on the carbonyl group of the peptide bond. An acyl-enzyme intermediate is formed, which is then liberated by hydrolysis. In the peptide-splicing reaction, a second peptide fragment appears to compete with water molecules for performing a nucleophilic attack on the acyl-enzyme intermediate, resulting in a transpeptidation reaction producing the spliced peptide. Experimental support for this model of reverse proteolysis includes evidence that the energy required to create the new peptide bond is recovered from the peptide bond that is cleaved at the amino-terminus of the excised fragment, and that the amino-terminus of the other fragment needs to be free for transpeptidation to occur.
2. Intermediate proteasome types
J. Abi Habib, E. De Plaen, B. Guillaume, N. Vigneron, A. Michaux
The proteasome exists in two forms: the standard proteasome, which is constitutively present in most cells, and the immunoproteasome, which is constitutive in many immune cells and can be induced by interferon-gamma in most other cells. They differ by the three catalytic subunits they use: ß1, ß2 and ß5 for the standard proteasome; ß1i, ß2i and ß5i for the immunoproteasome. We have described two new proteasome subtypes that are intermediate between the standard proteasome and the immunoproteasome (5). They contain only one (ß5i) or two (ß1i and ß5i) of the three inducible catalytic subunits of the immunoproteasome. These intermediate proteasomes represent 30 to 54% of the proteasome content of human liver, colon, small intestine and kidney. They are also present in human tumor cells and dendritic cells. They uniquely process several tumor antigens (5, 6). We are currently studying the function of these intermediate proteasomes, not only in terms of processing of antigenic peptides, but also for other functional aspects in which the proteasome plays a crucial role, such as the regulation of the cell cycle, the activation of transcription factors and the regulation of inflammation and immune responses.
3. Other proteases involved in antigen processing
N. Vigneron, A. Michaux, V. Stroobant
We are interested in characterizing the processing of human antigenic peptides that are not produced by the proteasome. We studied a proteasome-independent peptide derived from tumor protein MAGE-A3, and identified insulin-degrading enzyme as the protease producing this peptide (7). Insulin-degrading enzyme is a cytosolic metallopeptidase not previously known to play a role in the antigen processing pathway. The parental protein MAGE-A3 appears to be degraded along two parallel pathways involving insulin-degrading enzyme or the proteasome, each pathway producing a distinct set of antigenic peptides presented by MHC class I molecules. We are currently studying the processing of other proteasome-independent peptides and aiming to identify the protease(s) involved.
4. TAP-independent antigenic peptides
N. Vigneron, V. Stroobant, L. Pilotte
Presentation of most peptides depends on TAP, which transports peptides from the cytosol to the endoplasmic reticulum. A number of viruses and tumor cells tend to reduce their TAP expression to escape immune recognition. Therefore, there is great interest in the potential therapeutic use of peptides that are still presented in the absence of TAP. We are studying several such tumor peptides derived from cytosolic proteins. We aim at characterizing their processing and identifying the alternative transporter in charge of their transfer from the cytosol to the endoplasmic reticulum.
5. Cross-presentation
W. Ma, N. Vigneron, in collaboration with P. Courtoy and P. Van Der Smissen
Class I and class II molecules of the Major Histocompatibility Complex (MHC) are responsible for the presentation of antigenic peptides derived from intracellular proteins or from engulfed exogenous proteins, respectively. As an exception to this rule, cross-presentation enables dendritic cells to present on their MHC class I molecules antigenic peptides derived from exogenous material, through a mechanism that remains unclear. Cross-presentation is essential to the activation of CD8+ T lymphocytes against antigens derived from tumors and from viruses that do not infect dendritic cells. It is particularly efficient with long peptides, which are used in cancer vaccines. We studied the mechanism of long-peptide cross-presentation using human dendritic cells and specific CTL clones against melanoma antigens gp100 and Melan-A/MART1. We found that long-peptide cross-presentation does not depend on the proteasome nor the TAP transporter, and therefore follows a vacuolar pathway. We also observed that it makes use of newly synthesized MHC class I molecules that are loaded with suboptimal peptides. These nascent MHC-I molecules appear to diverge from the classical secretion pathway at an early stage and reach the late endosomes, where they exchange their suboptimal peptide cargo for the cross-presented peptide before reaching the cell surface in an endoH-sensitive form. These results indicate an alternative secretion pathway followed by HLA-I molecules that are used for cross-presentation, and may have implications for the development of vaccines based on long peptides.
II. MECHANISMS OF TUMORAL IMMUNE RESISTANCE (Group Jingjing Zhu)
1. Indoleamine 2,3-dioxygenase
M. Hennequart, J. Lamy, E. De Plaen, L. Pilotte, V. Stroobant, D. Colau
We previously discovered that tumors often resist immune rejection by expressing Indoleamine 2,3-dioxygenase (IDO), a tryptophan-degrading enzyme that is profoundly immunosuppressive (8). We showed that immune rejection was restored by administration of a pharmacological inhibitor of IDO. In collaboration with medicinal chemists in Namur and Lausanne, we identified several families of new IDO inhibitors that will be further optimized to develop drug candidates. We currently pursue functional studies on the mechanisms of IDO-induced immunosuppression, and on the signaling pathway responsible for IDO expression in tumors.
2. Tryptophan-dioxygenase
F. Schramme, D. Hoffmann, S. Klaessens, L. Pilotte, J. Lamy, E. De Plaen, V. Stroobant, D. Colau
Besides IDO, we recently uncovered the role of tryptophan-dioxygenase (TDO) in tumoral immune resistance (9). TDO is an unrelated tryptophan-degrading enzyme, which is highly expressed in the liver to regulate systemic tryptophan levels. We found TDO to be expressed in a high proportion of human tumors. We showed that TDO-expressing mouse tumors are no longer rejected by immunized mice. Moreover, we developed a TDO inhibitor, which, upon systemic treatment, restored the ability of mice to reject tumors (9). These results describe a mechanism of tumoral immune resistance based on TDO expression and establish proof-of-concept for the use of TDO inhibitors in cancer therapy. In April 2012, we have launched an LICR spin-off company, iTeos Therapeutics, which will develop inhibitors of IDO and TDO.
3. Transgenic mice developing autochthonous melanomas expressing P1A
C. Powis de Tenbossche, S. Cané, J. Zhu, C. Uyttenhove, N. Arts, E. De Plaen in collaboration with J. Van Snick
We have created a mouse model of autochthonous inducible melanoma expressing a defined tumor antigen (TIRP10B) (10). In this model, melanomas are induced (70% incidence) with tamoxifen, which, by activating CreER in melanocytes, induces the expression of Ha-Ras, the deletion of INK4a/ARF and the expression the tumor antigen encoded by cancer/germline gene P1A. A unique feature of this model is that melanomas first develop as non-aggressive highly pigmented tumors (Mela), which later dedifferentiate into unpigmented highly aggressive inflammatory tumors (Amela). We found that TGF-ß was a key factor responsible for this switch to aggressive tumors, which is reminiscent of the epithelial-to-mesenchymal transition (EMT) described in other contexts. We developed antibodies able to neutralize TGFß1 and TGFß3, and found that the former were able to increase survival of mice in this melanoma model. These results support the use of TGFß neutralizing therapies in the treatment of human melanoma.
The loss of pigmentation in aggressive tumors appears to result from the strong inflammation, and we identified a microRNA that is induced by interleukin-1 and downregulates expression of MITF, a transcription factor acting as a master regulator of pigmentation.
In this model, both pigmented (Mela) and unpigmented (Amela) tumors express the tumor antigen encoded by P1A. Mela tumors are ignored by the immune system, while Amela tumors are infiltrated by T lymphocytes that are rendered ineffective. We are studying the mechanisms responsible for this ineffectiveness. Our current results indicate that the tumor microenvironment actively induces the apoptosis of tumor-specific T lymphocytes that infiltrate the tumor. We are studying the molecular mechanisms responsible for this apoptosis.
References
J Immunother Cancer. 2021; 9(9):e003218.
Proc Natl Acad Sci U S A. 2021; 118(23):e2022447118.
J Immunother Cancer. 2021; 9(2):e001798.
Front Immunol. 2020; 11:601759.
Sci Rep. 2020; 10(1):15765.
Nat Commun. 2017; 8(1):1404.
J Immunol. 2016; 196(4):1711-20.
Proc Natl Acad Sci USA. 2012; 109(7):2497-502.
Dalet A, Robbins PF, Stroobant V, Vigneron N, Li YF, El-Gamil M, Hanada K, Yang JC, Rosenberg SA, Van den Eynde BJ.
Proc Natl Acad Sci USA. 2011; 108(29):E323-31.
Guillaume B, Chapiro J, Stroobant V, Colau D, Van Holle B, Parvizi G, Bousquet-Dubouch MP, Théate I, Parmentier N, Van den Eynde BJ.
Proc Natl Acad Sci U S A. 2010; 107(43):18599-604.
Nat Immunol. 2010; 11(5):449-54.
Warren EH, Vigneron NJ, Gavin MA, Coulie PG, Stroobant V, Dalet A, Tykodi SS, Xuereb SM, Mito JK, Riddell SR, Van den Eynde BJ.
Science. 2006; 313(5792):1444-7.
Huijbers IJ, Krimpenfort P, Chomez P, van der Valk MA, Song JY, Inderberg-Suso EM, Schmitt-Verhulst AM, Berns A, Van den Eynde BJ.
Cancer Res. 2006; 66(6):3278-86.
Vigneron N, Stroobant V, Chapiro J, Ooms A, Degiovanni G, Morel S, van der Bruggen P, Boon T, Van den Eynde BJ.
Science. 2004; 304(5670):587-90.
Nat Med. 2003; 9(10):1269-74.



 Download
Download
KANKERIMMUNOLOGIE EN IMMUNOTHERAPIE
