Volgt ons 

![]()
Het overkoepelende doel van ons onderzoek is het ontdekken van genetische varianten die menselijke ziektes veroorzaken, en te begrijpen hoe en waarom ze dat doen. We bestuderen verschillende ontwikkelingsstoornissen van vasculaire en skeletsystemen, en van bepaalde kankervormen. Van deze stoornissen zijn de moleculaire mechanismes vaak niet bekend. Huidige behandelmethodes zijn daarom breed en gericht op het verlichten van symptomen. Het identificeren van primaire oorzaken en bepalende factoren zou de ontwikkeling van effectievere, minder belastende behandelingen mogelijk maken. We gebruiken geavanceerde genetische technieken en stalen van patiënten om te ontdekken welke genen een rol spelen bij de ziektes. Vervolgens maken we cellulaire en dierlijke modellen waarmee we de moleculaire en biologische processen bij de ziektes verder analyseren en nieuwe therapieën testen. Al ons onderzoek begint met stalen van patiënten, en ons doel is de cirkel rond te maken: van het bed van de patiënt naar de labtafel en weer terug naar het bed.
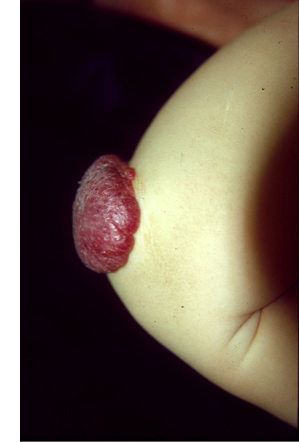
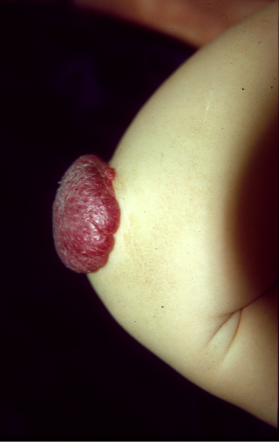
De bloed- en lymfevaatstelsels zijn cruciaal in de ontwikkeling, overleving en het functioneren van de mens. In een complex proces wordt een netwerk van slagaders, aders en haarvaten en lymfatische aders gevormd. Een grote reeks moleculen zorgt dat de vorming op gang komt en op het goede moment en de goede plek plaatsvindt. Een verstoord evenwicht tijdens dit proces resulteert in lokale constructiefouten, die vasculaire afwijkingen worden genoemd. Deze worden onderverdeeld in het type vaten dat ze beïnvloeden. De International Society for the study of Vascular Anomalies (ISSVA) heeft recent de classificatie uitgewerkt. De onbalansen kunnen ontstaan door mutaties in genen die coderen voor eiwitten die de ontwikkeling van vaten aansturen. Wij hebben genmutaties geïdentificeerd die verantwoordelijk zijn voor verschillende vasculaire afwijkingen, waaronder veneuze en lymfatische misvorming, lymfoedeem, en een combinatie van capillaire en slagaderlijke misvorming. Erfelijke vormen van de afwijkingen zijn zeldzaam. Hun niet-erfelijke tegenhangers komen veel vaker voor. Ze worden in het algemeen veroorzaakt door mutaties die alleen in de getroffen (misvormde) gebieden aanwezig zijn en niet worden overgedragen naar de volgende generatie.
De genmutaties die vasculaire afwijkingen veroorzaken, doen dit bijna altijd door de aantasting van endotheelcellen die de binnenwand van de bloedvaten vormen. We hebben aanzienlijke vooruitgang geboekt in het begrijpen van de veranderingen in deze cellen, en hoe ze kunnen worden gecorrigeerd. Klinische proeven op basis van deze gegevens zijn gestart bij het Centrum voor Vasculaire Afwijkingen.
Nuttige links
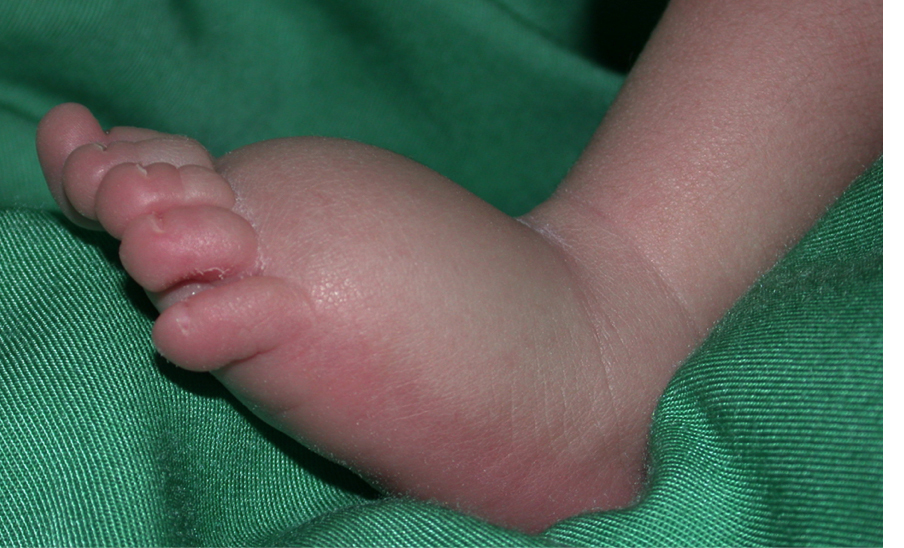
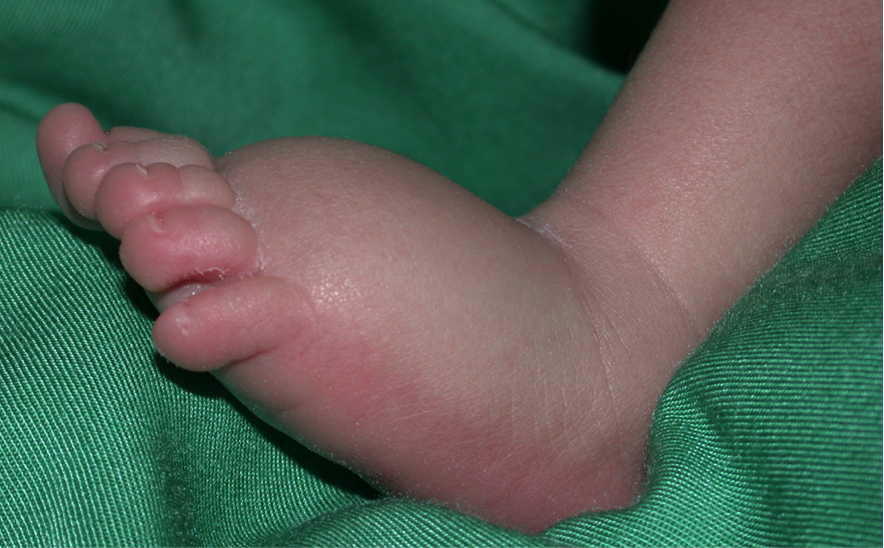
Lymfoedeem is een sterk invaliderende chronische ziekte als gevolg van een abnormale ontwikkeling en/of werking van het lymfatische systeem. Lymfevloeistof hoopt zich op, meestal in de onderste ledematen. Het leidt tot opzwellingen, fibrose en vatbaarheid voor secundaire infecties. Het functioneren van het getroffen lichaamsdeel is vaak beperkt. In Europa lijden meer dan een miljoen mensen aan lymfoedeem. De behandeling is beperkt tot herhaalde manuele lymfedrainage en het gebruik van compressieverbanden. In sommige gevallen kan een operatie verbetering geven, maar een genezende behandeling bestaat niet.
Lymfoedeem is onderverdeeld in primair (met een onbekende oorzaak) en secundair (met een bekende omgevingsoorzaak, bijvoorbeeld na een infectie of de behandeling van kanker). Door het bestuderen van getroffen gezinnen hebben we een aantal ziekte veroorzakende genen geïdentificeerd, en aangetoond dat mutaties dominant, recessief of zelfs de novo kunnen zijn. Sommige mutaties veroorzaken een veel bredere foetale lymfatische disfunctie (hydrops foetalis), wat het aantal diagnostische aanwijzingen vergroot. Tot op heden zijn al 28 genen bekend die primair lymfoedeem of aanleg voor de secundaire vorm veroorzaken. Deze verklaren echter de ziekte in minder dan een derde van de patiënten. Elk gemuteerd gen afzonderlijk verklaart slechts een klein percentage van de gevallen.
Nuttige links
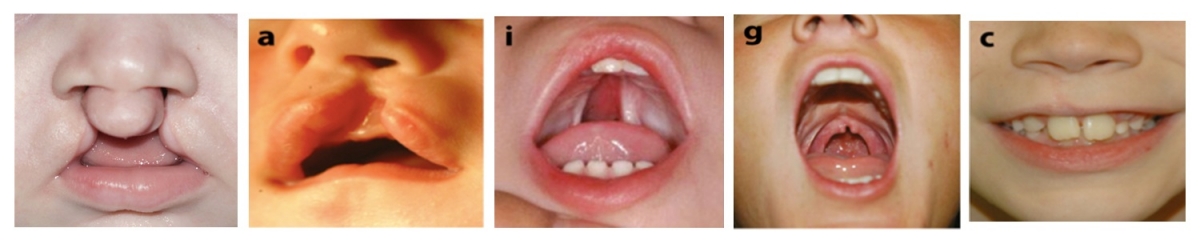
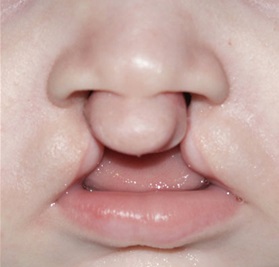
Een gespleten lip of gehemelte (CL/P) is de meest voorkomende aangeboren cranio-faciale afwijking. Net als andere veelvoorkomende menselijke aandoeningen, is CL /P een complexe ziekte, veroorzaakt door een combinatie van verschillende genvarianten en milieufactoren. Elke genetische variant draagt bij aan de aanleg voor de ziekte maar geen enkele werkt alleen, behalve in het geval van zeldzame, erfelijke vormen van CL/P. Dit maakt de identificatie van ziektegenen uitdagend. We beginnen derhalve met het identificeren van enkelvoudige genmutaties die verantwoordelijk zijn voor erfelijke CL/P. We scannen hiertoe de genomen van verschillende familieleden om varianten te identificeren die in alle getroffen individuen voorkomen. We testen vervolgens dezelfde genen in niet-erfelijke CL/P, op varianten die significant vaker of minder vaak voorkomen bij patiënten dan bij gezonde individuen. Dit kan aangeven dat ze een predisponerend of beschermend effect uitoefenen. CL/P kan samen gaan met andere afwijkingen in verschillende syndromen. We hebben ontdekt dat genen die bepaalde CL/P-inclusieve syndromen veroorzaken ook bijdragen aan geïsoleerde CL/P gevallen als ze minder gemuteerd zijn. Dit heeft ons een extra middel gegeven om mogelijke ziektegenen te identificeren.
In samenwerking met andere onderzoeksgroepen, richten wij ons op de identificatie van genmutaties in verschillende vormen van kanker: neuro-endocriene en hersentumoren, borstkanker en hematologische maligniteiten. We zoeken zowel naar mutaties die de ziekte initiëren als mutaties die de progressie beïnvloeden. Daarnaast proberen we merkers te vinden die helpen bij de diagnose en prognose. Dit kunnen genetische veranderingen zijn (voor een "moleculaire" diagnose of classificatie), maar ook eiwitten gedetecteerd in bloedstalen of met biopsie verkregen weefselstalen.
Met Prof. D. MANICOURT
We proberen de gen(en) te identificeren die verantwoordelijk zijn voor het Ehlers-Danlos syndroom (EDS), type hypermobiliteit, een weinig bekende en toch relatief frequente aandoening. Het syndroom is niet te genezen en er is geen afwijkend collageen gevonden. Vanwege hun uitzonderlijke beweeglijkheid, zijn gewrichten gevoelig voor dislocaties en sublocaties.
Het EDS-syndroom is een veel voorkomende oorzaak van ernstige, vroege en multifocale osteoarthritis. Andere verschijnselen zijn onder meer chronische pijn, blauwe plekken, darmstoornissen, dysautonomie en aortawortel dilatatie.

 Het gebied van de menselijke genetica is in een versnelling gekomen door massale parallelle sequencering ("Next Generation Sequencing" of "NGS"). Grote hoeveelheden data worden geproduceerd met toenemende snelheid. De technologie genereert een aanzienlijk aantal valse positieven en het onderscheiden van sequencering fouten van echte mutaties is geen eenvoudige taak. Vervolgens moeten de interessante veranderingen worden onderscheiden tussen de tienduizenden varianten. Dit vereist de annotatie van informatie over de veranderingen uit verschillende bronnen, evenals geavanceerde filtermethodes. Wij huisvesten het Technisch Platform voor Genetica van de Faculteit der Geneeskunde van de Université catholique de Louvain, zodat onze wetenschappers kunnen beschikken over de nieuwste DNA-sequencering technologieën. Wij nemen deel aan regionale en nationale ontwikkeling van tools voor data-analyse. We voorzien ook ondersteuning op het gebied van bioinformatica, die essentieel is voor de analyse van NGS data. Verder hebben we Highlander ontwikkeld, een softwarepakket dat een aantal in-silico analyseprogramma's en hulpmiddelen integreert met een gebruiksvriendelijke grafische interface. Dit vergroot onze mogelijkheden om de genetische en epigenetische bases van ziektes te identificeren en te verkennen. We ontwikkelen ook een uitgebreid LIMS (Laboratory Information Management System) voor het efficiënt beheren van gegevens uit onze biobanken, zodat we monsters, patiënten, klinische informatie, experimenten en publicaties kunnen koppelen. Omdat bio-informatica steeds belangrijker wordt voor genetisch onderzoek, zijn andere in-silico projecten in de toekomst voorzien.
Het gebied van de menselijke genetica is in een versnelling gekomen door massale parallelle sequencering ("Next Generation Sequencing" of "NGS"). Grote hoeveelheden data worden geproduceerd met toenemende snelheid. De technologie genereert een aanzienlijk aantal valse positieven en het onderscheiden van sequencering fouten van echte mutaties is geen eenvoudige taak. Vervolgens moeten de interessante veranderingen worden onderscheiden tussen de tienduizenden varianten. Dit vereist de annotatie van informatie over de veranderingen uit verschillende bronnen, evenals geavanceerde filtermethodes. Wij huisvesten het Technisch Platform voor Genetica van de Faculteit der Geneeskunde van de Université catholique de Louvain, zodat onze wetenschappers kunnen beschikken over de nieuwste DNA-sequencering technologieën. Wij nemen deel aan regionale en nationale ontwikkeling van tools voor data-analyse. We voorzien ook ondersteuning op het gebied van bioinformatica, die essentieel is voor de analyse van NGS data. Verder hebben we Highlander ontwikkeld, een softwarepakket dat een aantal in-silico analyseprogramma's en hulpmiddelen integreert met een gebruiksvriendelijke grafische interface. Dit vergroot onze mogelijkheden om de genetische en epigenetische bases van ziektes te identificeren en te verkennen. We ontwikkelen ook een uitgebreid LIMS (Laboratory Information Management System) voor het efficiënt beheren van gegevens uit onze biobanken, zodat we monsters, patiënten, klinische informatie, experimenten en publicaties kunnen koppelen. Omdat bio-informatica steeds belangrijker wordt voor genetisch onderzoek, zijn andere in-silico projecten in de toekomst voorzien.
The Human Molecular Genetics group studies the molecular-genetic bases of a variety of developmental disorders including vascular anomalies and cleft lip and palate, hypermobility, as well as cancers, in order to improve their diagnosis and clinical management, and uncover new targets for therapy. Our laboratory combines a large bio-bank of patient blood and tissue samples associated with extensive clinical data, with large-scale genotyping data generated using cutting-edge Next Generation Sequencing techniques and a state-of-the-art data analysis pipeline developed in-house. Gene-discovery is followed by the generation of cell and animal models in which disruption of the culprit gene is used to recapitulate features of the human disease. These models are then used to study the molecular and biological consequences of disease causative genetic changes, as well as to test potential therapeutic solutions. All of our research begins with samples obtained from patients, and we aim to go full circle: from bed to bench-side back to bed, providing patients with viable and tangible knowledge-based improvements in their health-care.
 |
 |
 |
| Vasculaire afwijkingen | Lymfoedeem | Gespleten lip of gehemelte |
|
|
|
| Kanker | Hypermobiliteit | Bioinformatics |
Nat Rev Dis Primers. 2021; 7(1):77.
Circ Res. 2021; 129(1):155-173.
Circ Res. 2021; 129(1):136-154.
JCI Insight. 2021; 6(15):e149831.
Hammer J, Seront E, Duez S, Dupont S, Van Damme A, Schmitz S, Hoyoux C, Chopinet C, Clapuyt P, Hammer F, Vikkula M, Boon LM.
Orphanet J Rare Dis. 2018; 13(1):191.
Quéré I, Nagot N, Vikkula M.
N Engl J Med. 2018; 378(21):2047-8.
Basha M, Demeer B, Revencu N, Helaers R, Theys S, Bou Saba S, Boute O, Devauchelle B, Francois G, Bayet B, Vikkula M.
J Med Genet. 2018; 55(7):449-58.
Amyere M, Revencu N, Helaers R, Pairet E, Baselga E, Cordisco M, Chung W, Dubois J, Lacour JP, Martorell L, Mazereeuw-Hautier J, Pyeritz RE, Amor DJ, Bisdorff A, Blei F, Bombei H, Dompmartin A, Brooks D, Dupont J, González-Enseñat MA, Frieden I, Gérard M, et al.
Circulation. 2017; 136(11):1037-48.
J Invest Dermatol. 2017; 137(1):207-16.
Am J Hum Genet. 2015; 97(6):914-21.
J Clin Invest. 2015; 125(9):3491-504.
Wassef M, Blei F, Adams D, Alomari A, Baselga E, Berenstein A, Burrows P, Frieden IJ, Garzon MC, Lopez-Gutierrez JC, Lord DJ, Mitchel S, Powell J, Prendiville J, Vikkula M; ISSVA Board and Scientific Committee.
Pediatrics. 2015; 136(1):e203-14.
Revencu N, Boon LM, Mendola A, Cordisco MR, Dubois J, Clapuyt P, Hammer F, Amor DJ, Irvine AD, Baselga E, Dompmartin A, Syed S, Martin-Santiago A, Ades L, Collins F, Smith J, Sandaradura S, Barrio VR, Burrows PE, Blei F, Cozzolino M, Brunetti-Pierri N, et al.
Hum Mutat. 2013; 34(12):1632-41.
Limaye N, Wouters V, Uebelhoer M, Tuominen M, Wirkkala R, Mulliken JB, Eklund L, Boon LM, Vikkula M.
Nat Genet. 2009; 41(1):118-24.
Jinnin M, Medici D, Park L, Limaye N, Liu Y, Boscolo E, Bischoff J, Vikkula M, Boye E, Olsen BR.
Nat Med. 2008; 14(11):1236-46.
Vikkula M, Boon LM, Carraway KL 3rd, Calvert JT, Diamonti AJ, Goumnerov B, Pasyk KA, Marchuk DA, Warman ML, Cantley LC, Mulliken JB, Olsen BR.
Cell. 1996; 87(7):1181-90.
Vikkula M, Mariman EC, Lui VC, Zhidkova NI, Tiller GE, Goldring MB, van Beersum SE, de Waal Malefijt MC, van den Hoogen FH, Ropers HH, et al.
Cell. 1995; 80(3):431-7.

 Download
Download
GENETISCHE BASES VAN MENSELIJKE ZIEKTES
