Follow us 

![]()
The overarching aim of our research is to discover the genetic variations that drive human disease, and to understand how and why they do so. We study a variety of developmental disorders of the cardiovascular and skeletal systems, as well as certain cancers. The molecular mechanisms of many are not known, and current treatments are therefore broad-based and aimed at alleviating symptoms. Identification of the primary causes as well as modulating factors would greatly facilitate the development of more effective, less caustic treatments. Towards this goal, we use cutting-edge genetic techniques on patient samples for gene-discovery, followed by the generation of cellular and animal models in which to further dissect disease processes and test new therapies. All of our research begins with samples obtained from patients, and we aim to go the full circle: from bed to bench-side and back to bed, providing patients with viable and tangible knowledge-based improvements in their health-care.
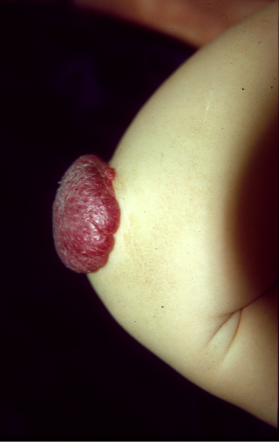
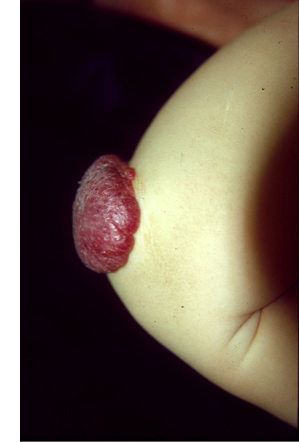 The blood and lymphatic vascular systems are crucial to the development, survival and function of the human organism. In an intricate process that relies on a large array of molecules to provide the right cues at the right times and in the right places, a well-organized network of arteries, veins, capillaries and lymphatic vessels is formed. Imbalances that occur during this process result in the formation of localized structural defects, collectively termed vascular anomalies, which are categorized by the type of vessel(s) they affect. International Society for the Study of Vascular Anomalies (ISSVA) has recently detailed their classification. Such imbalances can be caused by mutations in genes encoding proteins that drive vascular development. We have identified gene mutations responsible for many different types of vascular anomalies including venous and lymphatic malformations, lymphedema, and capillary malformation in combination with arteriovenous malformation. In general, familial forms of these anomalies are very rare, and are caused by germline mutations. Their sporadic counterparts are far more common, and generally caused by somatic mutations, i.e., mutations that are present only in the affected (malformed) areas and therefore, not transmitted from one generation to the next. The gene mutations that cause vascular anomalies almost always affect endothelial cells that form the inner lining of blood vessels, and we have made considerable progress in understanding the changes that they cause in these cells, and how they can be corrected. Clinical trials based on these data have been initiated at Centre for Vascular Anomalies.
The blood and lymphatic vascular systems are crucial to the development, survival and function of the human organism. In an intricate process that relies on a large array of molecules to provide the right cues at the right times and in the right places, a well-organized network of arteries, veins, capillaries and lymphatic vessels is formed. Imbalances that occur during this process result in the formation of localized structural defects, collectively termed vascular anomalies, which are categorized by the type of vessel(s) they affect. International Society for the Study of Vascular Anomalies (ISSVA) has recently detailed their classification. Such imbalances can be caused by mutations in genes encoding proteins that drive vascular development. We have identified gene mutations responsible for many different types of vascular anomalies including venous and lymphatic malformations, lymphedema, and capillary malformation in combination with arteriovenous malformation. In general, familial forms of these anomalies are very rare, and are caused by germline mutations. Their sporadic counterparts are far more common, and generally caused by somatic mutations, i.e., mutations that are present only in the affected (malformed) areas and therefore, not transmitted from one generation to the next. The gene mutations that cause vascular anomalies almost always affect endothelial cells that form the inner lining of blood vessels, and we have made considerable progress in understanding the changes that they cause in these cells, and how they can be corrected. Clinical trials based on these data have been initiated at Centre for Vascular Anomalies.
Useful links
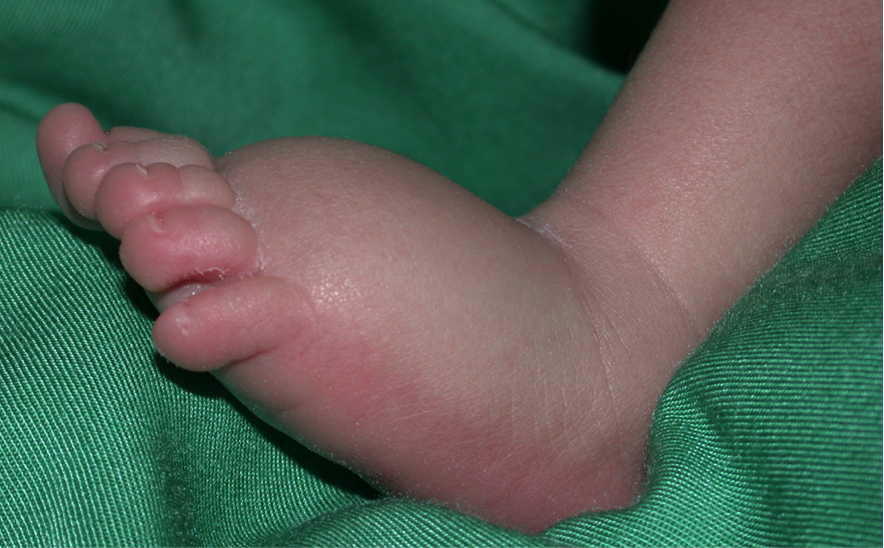
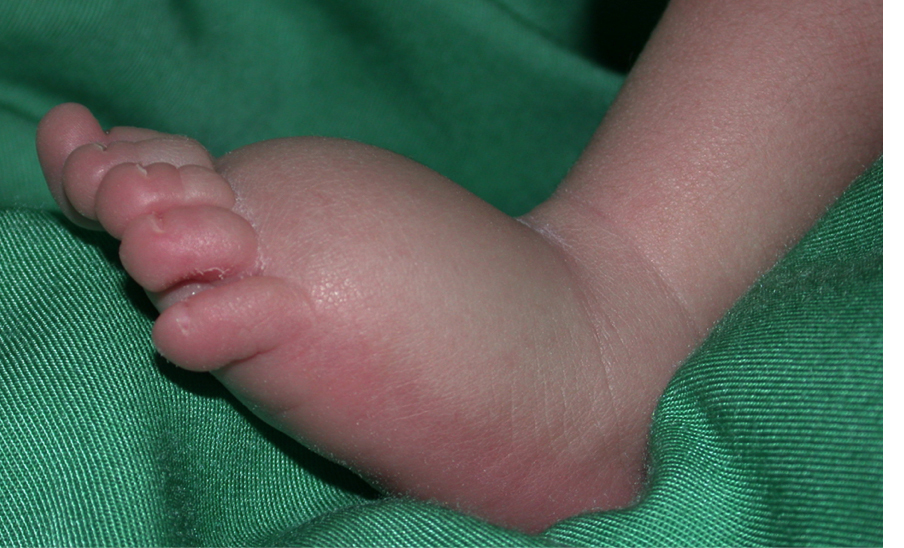 Lymphedema is a strongly invalidating chronic disease resulting from abnormal development and/or function of the lymphatic system. Lymph is not drained from interstitial tissues, but accumulates, most often in lower extremities, causing enlargement, fibrosis and predisposition to secondary infections. Function of the affected body part is often compromised. In Europe, over a million people are affected. Therapy is limited to repeated manual lymphatic drainage, and use of compressive garments. In some cases, surgery may be helpful, but no curative therapy exists.
Lymphedema is a strongly invalidating chronic disease resulting from abnormal development and/or function of the lymphatic system. Lymph is not drained from interstitial tissues, but accumulates, most often in lower extremities, causing enlargement, fibrosis and predisposition to secondary infections. Function of the affected body part is often compromised. In Europe, over a million people are affected. Therapy is limited to repeated manual lymphatic drainage, and use of compressive garments. In some cases, surgery may be helpful, but no curative therapy exists.
Lymphedema is divided into primary (unknown cause) and secondary (known environmental cause, such as after infection or cancer therapy). Primary lymphedema is sometimes inherited from generation to generation, similar to vascular anomalies. By studying such families, we have several disease-causative genes for primary lymphedema, and demonstrated that mutations can be dominant, recessive or even de novo. Some cause a much wider fetal lymphatic dysfunction (hydrops fetalis), or syndromes, enlarging diagnostic testing indications. To date, 28 genes are already known to cause primary lymphedema and/or predispose to the secondary form, but these account only for less than a third of the samples, each gene explaining a small percentage of the cases.
 |
Useful links
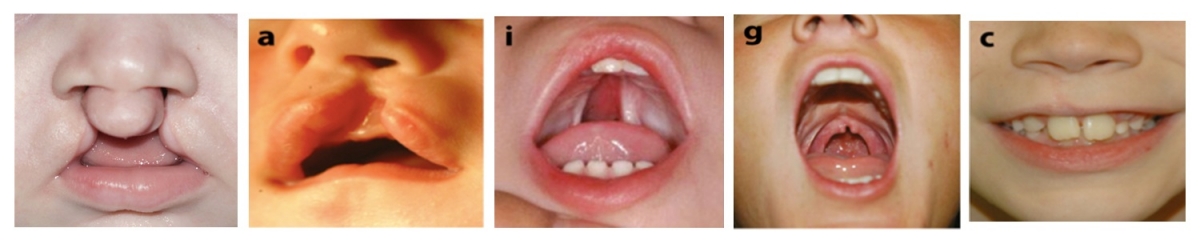
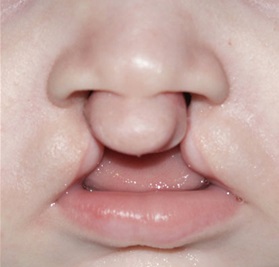
Cleft lip and/or palate (CL/P) is the most common congenital craniofacial anomaly. Like many other common human conditions including hypertension and diabetes, CL/P is a complex disease caused by the combination of several different gene variants in concert with environmental factors. Each genetic variant contributes to disease predisposition, but none acts alone, except in the case of rare, familial forms of CL/P. In addition, different combinations of genes cause disease in different individuals. This makes the identification of disease genes challenging.
We therefore begin by identifying single gene mutations that are responsible for inherited isolated CL/P, by studying the genomes of several family members to identify variants that are present in all affected individuals i.e., that co-segregate with the disease. We then test these same genes in sporadic CL/P, for variants that occur significantly more or less often in patients than in healthy individuals, indicating they may exert a predisposing or protective effect.
CL/P can occur with other abnormalities in various syndromes. We discovered that genes that cause certain syndromic forms of CL/P also contribute to isolated CL/P when less severely mutated. This has given us additional means by which to identify candidate disease genes.
In collaboration with other research groups, we aim to identify driver and modifier mutations (i.e., mutations that initiate disease vs. those that affect progression) in a variety of cancers: neuroendocrine and cerebral tumors, breast cancer, and hematological malignancies. In addition, we seek to find markers that will aid in diagnosis and prognostication. These include genetic changes (for a “molecular” diagnosis or classification), as well as proteins detected in the circulation or in biopsied tissue samples.
With Prof. D. MANICOURT
We try to identify the gene(s) responsible for the Ehlers-Danlos syndrome (EDS), hypermobility type, an under-recognized and yet relatively frequent condition. The syndrome has no cure and no distinctive biochemical collagen findings. Because of their excessive range of movement, joints are prone to joint dislocation and subluxations. The syndrome is a common cause of severe, early and multifocal osteoarthritis. Other clinical manifestations include chronic pain, easy bruising, functional bowel disorders, autonomic dysfunction and aortic root dilatation.


The field of human genetics is being revolutionized by massive parallel sequencing (“Next Generation Sequencing” or “NGS”). Large amounts of data are being produced at ever-increasing rates. Targeted exome sequencing can be completed in a few days using NGS, allowing for new variant discovery in a matter of weeks. The technology generates considerable numbers of false positives, and the differentiation of sequencing errors from true mutations is not a straightforward task. Moreover, the identification of changes-of-interest from amongst tens of thousands of variants requires annotation drawn from various sources, as well as advanced filtering capabilities. We host the Genetics technical platform of the Faculty of Medicine of the Université catholique de Louvain, making those latest DNA sequencing technologies available to our scientists. We participate in regional and national development of analysis pipelines. We also provide bioinformatics support, essential for the analysis of NGS data. Furthermore, we have developed Highlander, a software package that integrates several in-silico analysis programs and utilities with a user-friendly graphical interface. This enhances our ability to identify and explore the genetic and epigenetic bases of disease. We are also developing a comprehensive LIMS (Laboratory Information Management System) that will allow us to efficiently manage data from our biobanks, linking samples, patients, clinical information, experiments, results and publications. As bioinformatics becomes an essential component of genetic research, other in-silico projects will be implemented in the future.
The Human Molecular Genetics group studies the molecular-genetic bases of a variety of developmental disorders including vascular anomalies and cleft lip and palate, hypermobility, as well as cancers, in order to improve their diagnosis and clinical management, and uncover new targets for therapy. Our laboratory combines a large bio-bank of patient blood and tissue samples associated with extensive clinical data, with large-scale genotyping data generated using cutting-edge Next Generation Sequencing techniques and a state-of-the-art data analysis pipeline developed in-house. Gene-discovery is followed by the generation of cell and animal models in which disruption of the culprit gene is used to recapitulate features of the human disease. These models are then used to study the molecular and biological consequences of disease causative genetic changes, as well as to test potential therapeutic solutions. All of our research begins with samples obtained from patients, and we aim to go full circle: from bed to bench-side back to bed, providing patients with viable and tangible knowledge-based improvements in their health-care.
 |
 |
 |
| Vascular anomalies | Lymphedema | Clef lip and Palate |
|
|
|
| Cancer | Hypermobility | Bioinformatics |
Nat Rev Dis Primers. 2021; 7(1):77.
Circ Res. 2021; 129(1):155-173.
Circ Res. 2021; 129(1):136-154.
JCI Insight. 2021; 6(15):e149831.
Hammer J, Seront E, Duez S, Dupont S, Van Damme A, Schmitz S, Hoyoux C, Chopinet C, Clapuyt P, Hammer F, Vikkula M, Boon LM.
Orphanet J Rare Dis. 2018; 13(1):191.
Quéré I, Nagot N, Vikkula M.
N Engl J Med. 2018; 378(21):2047-8.
Basha M, Demeer B, Revencu N, Helaers R, Theys S, Bou Saba S, Boute O, Devauchelle B, Francois G, Bayet B, Vikkula M.
J Med Genet. 2018; 55(7):449-58.
Amyere M, Revencu N, Helaers R, Pairet E, Baselga E, Cordisco M, Chung W, Dubois J, Lacour JP, Martorell L, Mazereeuw-Hautier J, Pyeritz RE, Amor DJ, Bisdorff A, Blei F, Bombei H, Dompmartin A, Brooks D, Dupont J, González-Enseñat MA, Frieden I, Gérard M, et al.
Circulation. 2017; 136(11):1037-48.
J Invest Dermatol. 2017; 137(1):207-16.
Am J Hum Genet. 2015; 97(6):914-21.
J Clin Invest. 2015; 125(9):3491-504.
Wassef M, Blei F, Adams D, Alomari A, Baselga E, Berenstein A, Burrows P, Frieden IJ, Garzon MC, Lopez-Gutierrez JC, Lord DJ, Mitchel S, Powell J, Prendiville J, Vikkula M; ISSVA Board and Scientific Committee.
Pediatrics. 2015; 136(1):e203-14.
Revencu N, Boon LM, Mendola A, Cordisco MR, Dubois J, Clapuyt P, Hammer F, Amor DJ, Irvine AD, Baselga E, Dompmartin A, Syed S, Martin-Santiago A, Ades L, Collins F, Smith J, Sandaradura S, Barrio VR, Burrows PE, Blei F, Cozzolino M, Brunetti-Pierri N, et al.
Hum Mutat. 2013; 34(12):1632-41.
Limaye N, Wouters V, Uebelhoer M, Tuominen M, Wirkkala R, Mulliken JB, Eklund L, Boon LM, Vikkula M.
Nat Genet. 2009; 41(1):118-24.
Jinnin M, Medici D, Park L, Limaye N, Liu Y, Boscolo E, Bischoff J, Vikkula M, Boye E, Olsen BR.
Nat Med. 2008; 14(11):1236-46.
Vikkula M, Boon LM, Carraway KL 3rd, Calvert JT, Diamonti AJ, Goumnerov B, Pasyk KA, Marchuk DA, Warman ML, Cantley LC, Mulliken JB, Olsen BR.
Cell. 1996; 87(7):1181-90.
Vikkula M, Mariman EC, Lui VC, Zhidkova NI, Tiller GE, Goldring MB, van Beersum SE, de Waal Malefijt MC, van den Hoogen FH, Ropers HH, et al.
Cell. 1995; 80(3):431-7.

 Download
Download GENETIC BASES OF HUMAN DISEASE
