In our laboratory, we are interested in understanding the mechanisms that are set up by cancer cells to acquire this “endless youthful” property that ensures indefinite cellular proliferation potential. Understanding these mechanisms is expected to help identifying new potential therapeutic targets in the fight against cancer.
Telomeres are specialized structures that protect chromosome ends. They can be viewed as “protective caps”. The attribution of Nobel Prize in Medicine 2009 to the discovery of telomeres and telomerase has reinforced the importance of studying telomeres, namely in the context of cancer research.
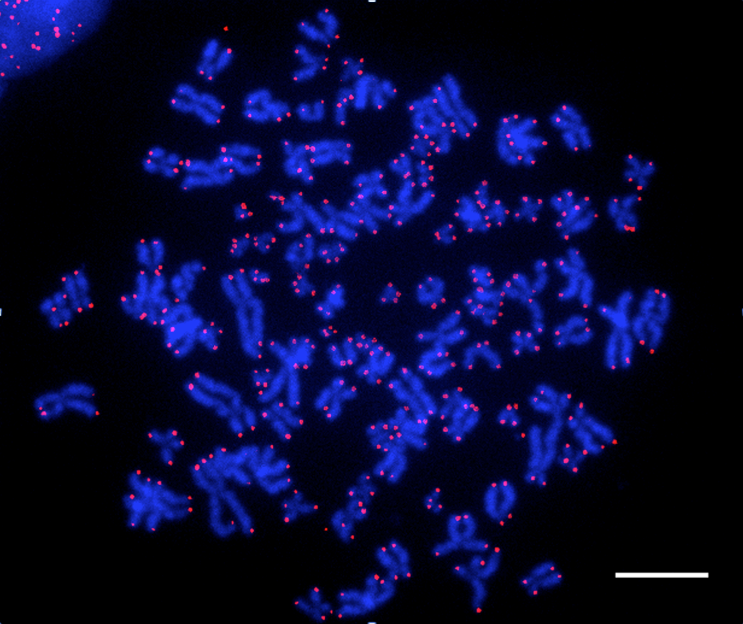
Legend:
Visualization of human telomeres with a fluorescent probe (FISH technique-Fluorescent In Situ Hybridization). Chromosomes are stained in blue. Telomeres appear as pink signals at chromosome ends.
The ability to maintain these “protective caps” is of major importance in dividing cells, as the natural process of cell division erodes chromosome ends. During the first embryonic divisions, cells are able to ensure the protection, but this ability is next lost in most of our cells, as they do not divide anymore. In this way, nature protects us against indefinite cellular proliferation that characterizes cancer cells. How, then, can tumors form if telomere maintenance is switched off in somatic cells? Acquisition of a telomere maintenance mechanism requires to bypass natural protective barriers against cancer, through mutations and chromosome rearrangements. A fraction of the cells that go through tumorigenesis manages to breach barriers and to set up such a mechanism, and these cells will be the only ones to proliferate in the body. In the laboratory, we are interested in understanding this and we would like to acquire a better knowledge of telomere maintenance mechanisms in order to identify new putative anti-cancer treatments.
Telomeres are specialized structures present at chromosome ends. They are made of telomeric DNA (TTAGGG repeats in vertebrates), proteins and non-coding RNA that, together with a proper heterochromatin (HC) structure, contribute to the maintenance of linear chromosome extremities. At healthy telomeres, binding of a specialized protein complex, called shelterin, together with the formation of a lariat structure, known as t-loop (telomeric-loop), suppress homologous recombination as well as end-to-end fusions (Figure 1).
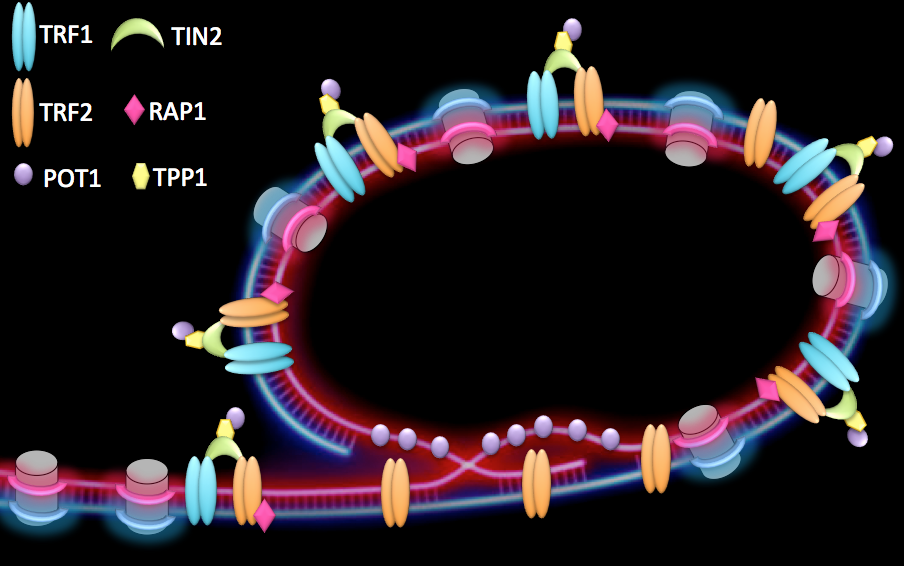
Figure 1:
Proposed conformation of human telomeres. Chromosome ends are believed to form a lariat structure, known as telomeric-loop, that protect them from unwanted activation of DNA damage response. Proteins from the shelterin complex bind telomeric DNA and are required for proper telomere function.
In the absence of a telomere maintenance mechanism, and due to the inability of DNA polymerase to fully replicate chromosome ends (the so-called “end-replication problem”), telomeres progressively shorten with cell divisions until they get too short to properly protect chromosome ends. Unprotected telomeres are then recognized as DNA damage signals by the cells; this activates p53 and triggers entry of cells into senescence. This scenario takes place during cellular ageing and contributes to impairment of organ function and organismal ageing. The inability of most of our cells to maintain telomeres is currently viewed as a major barrier against tumor development. How do cancer cells acquire immortality then? During the process of tumorigenesis, cells need to bypass the senescence barrier and to activate a telomere maintenance mechanism. In about 90% of tumours, this is achieved through reactivation of telomerase enzyme, while the other 10% activate a homologous recombination-based mechanism, called ALT (for Alternative Lengthening of Telomeres). Although ALT tumors contribute to only 10% of total cancers, they are frequently detected in sarcomas, glioblastomas and astrocytomas, with a high prevalence in pediatric tumours (Figure 2).
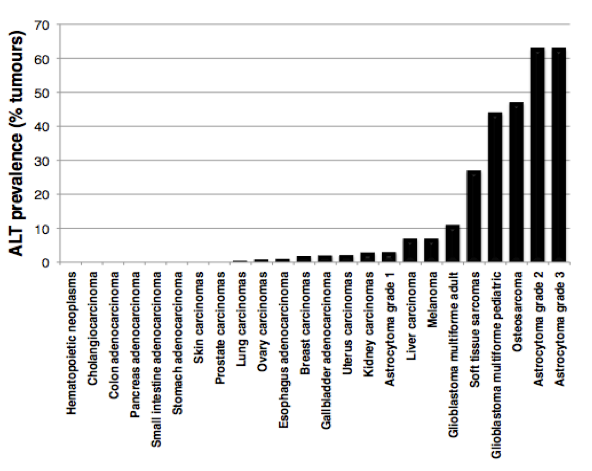
Figure 2:
ALT prevalence in human tumors. The graph is based on data published in Heaphy et al., Am J Pathol, 2011.
ALT cells are characterized by telomere length heterogeneity, extrachromosomal telomeric DNA fragments, frequent telomeric sister chromatid exchanges and co-localization of telomeric DNA with specialized PML bodies (ALT-associated PML bodies or APBs) (Figure 3A-B). More recently, it was also found that ALT telomeres are characterized by the presence of distinct telomeric repeats that diverge from the canonical TTAGGG and allow the binding of a class of proteins, known as orphan receptors (Figure 3C). These nuclear receptors presumably compete with canonical telomere-binding proteins but how their presence at telomeres contributes to ALT mechanism is still unknown.
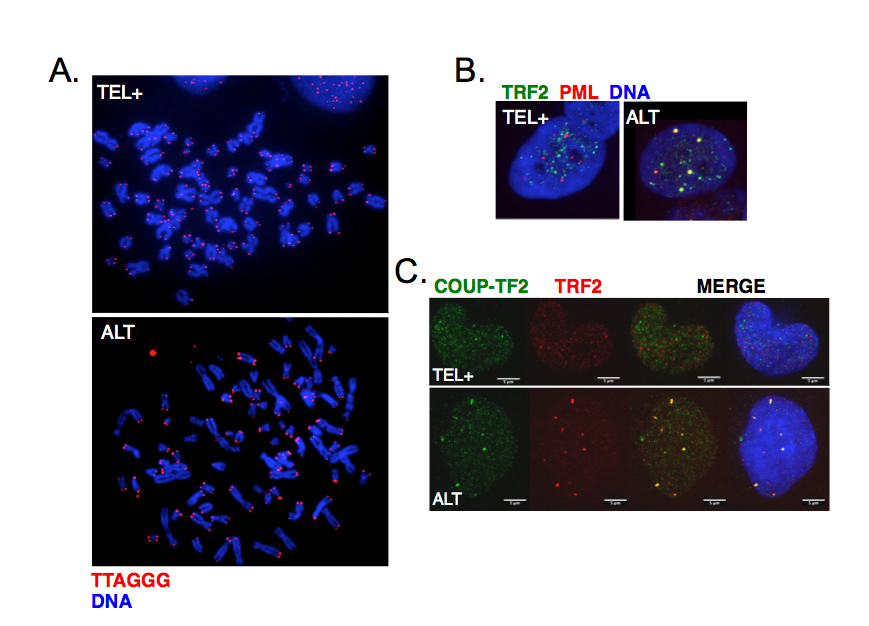
Figure 3:
Main characteristics of ALT cells. A. Telomeres were detected by FISH using a Cy3-(CCCTAA)3 PNA probe in either telomerase-positive (TEL+) or ALT human cells. Chromosomes were stained with DAPI. The heterogeneity of telomeric signals is clearly visible in ALT cells. B. Co-localization of telomeres with PML bodies in ALT cells visualized by immunofluorescence using antibodies against TRF2 shelterin protein (green) and PML (red). C. Binding of COUP-TF2 orphan receptor at ALT telomeres detected by immunofluorescence using antibodies against TRF2 shelterin protein (red) and COUP-TF2 (green).
To date, we still do not have a clear view of the cellular alterations that contribute to activation of homologous recombination at telomeres during the establishment of ALT. In the context of anti-cancer therapy targeting telomere maintenance, it would however be useful to find ALT-specific targets. This, not only for ALT tumor eradication, but also in the likely event of ALT survivor emergence following anti-telomerase therapy. Hence, in the laboratory, we are interested in understanding the mechanisms of ALT activation. We previously showed that subtelomeric DNA hypomethylation is not required to activate ALT (Tilman et al., 2009) and more recently, we established new tools for the identification of genetic and/or epigenetic ALT-specific alterations in immortalized human cell lines (Van Beneden et al., submitted).
The second research axis in the lab focuses on the study of telomeric non-coding RNAs. Despite their heterochromatic structure, telomeres are indeed transcribed into non-coding RNAs, dubbed TERRA for Telomeric Repeat-containing RNA. TERRA molecules remain partly associated with telomeres (Figure 4) where they are believed to fulfill important functions, including telomeric heterochromatin formation, regulation of telomerase access to telomeres or t-loop formation. Agreeing with multiple roles of TERRA in various biological processes, these non-coding RNA species interact with a large variety of proteins (Scheibe et al., 2013). We recently showed that the abundance of human TERRA molecules is regulated by telomere length (Arnoult et al., 2012, Van Beneden et al., 2013) and we now wish to pursue our study of telomere transcription regulation, both in human cells and in mouse. We would also like to understand more about the roles of TERRA, not only in the context of cancer cells, but also in more physiological conditions. Are telomeres transcribed in all tissues? Are TERRA molecules required for normal cell viability? Is telomere expression restricted to dividing cells?
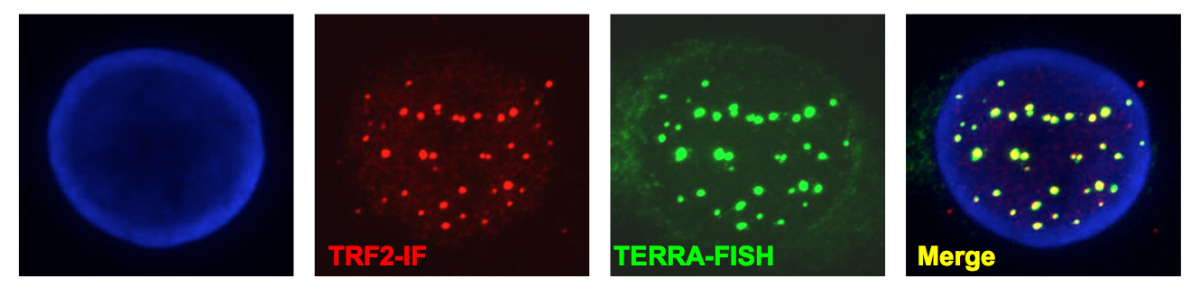
Figure 4:
Telomeric transcripts at human telomeres. Telomeres are transcribed into non-coding RNA molecules that remain partly associated with telomeres. Telomeric transcripts, dubbed TERRA (Telomeric Repeat-containing RNA) were detected by RNA-FISH using a FAM-(CCCTAA)7 LNA probe (green). Telomeres were visualized by immunofluorescence using antibodies against TRF2 shelterin protein (red) and chromosomes were stained with DAPI.
Bioessays. 2021; e2100099.
EMBO J. 2021; 40(6):e106336.
RNA. 2021; 27(1):106-121.
Aging. 2020; 12(20):19911-19922.
Episkopou H, Diman A, Claude E, Viceconte N, Decottignies A.
Mol Cell. 2019; 75(3):469-82.e6.
Benyelles M, Episkopou H, O'Donohue MF, Kermasson L, Frange P, Poulain F, Burcu Belen F, Polat M, Bole-Feysot C, Langa-Vives F, Gleizes PE, de Villartay JP, Callebaut I, Decottignies A, Revy P.
EMBO Mol Med. 2019; 11(7):e10201.
Diman A, Decottignies A.
FEBS J. 2018; 285(8):1389-98.
Viceconte N, Dheur MS, Majerova E, Pierreux CE, Baurain JF, van Baren N, Decottignies A.
Cell Rep. 2017; 19(12):2529-43.
Diman A, Boros J, Poulain F, Rodriguez J, Purnelle M, Episkopou H, Bertrand L, Francaux M, Deldicque L, Decottignies A.
Sci Adv. 2016; 2(7):e1600031.
Boros J, Arnoult N, Stroobant V, Collet JF, Decottignies A.
Mol Cell Biol. 2014; 34(19):3662-74.
Episkopou H, Draskovic I, Van Beneden A, Tilman G, Mattiussi M, Gobin M, Arnoult N, Londoño-Vallejo A, Decottignies A.
Nucleic Acids Res. 2014; 42(7):4391-405.
Scheibe M, Arnoult N, Kappei D, Buchholz F, Decottignies A, Butter F, Mann M.
Genome Res. 2013; 23(12):2149-57.
Arnoult N, Van Beneden A, Decottignies A.
Nat Struct Mol Biol. 2012; 19(9):948-56.
Tilman G, Arnoult N, Lenglez S, Van Beneden A, Loriot A, De Smet C, Decottignies A.
Epigenetics. 2012; 7(8):903-13.
Mattiussi M, Tilman G, Lenglez S, Decottignies A.
Cell Signal. 2012; 24(3):708-17.
Mol Biomed. 2021; 2(32):1-16

 Download
Download TELOMERES & EPIGENETICS
