26/04/2019
Liver cancer, more specifically hepatocellular carcinoma (HCC) - a tumor initiated in hepatocytes-, is the third–most common cause of cancer death worldwide. It results from a variety of conditions such as viral infections or cirrhosis, thereby leading to significant heterogeneity among patients and variable response to therapeutic agents. As a consequence, there is need to develop therapies that are specific to individual patients.
The disease is associated with the concomitant deregulation of several genes that are functionally organised as networks. It is the network as a whole, not an individual gene, which impacts cancer progression. How a specific network is affected varies from one patient to the other, prompting the need to design tools that can predict the behaviour of gene networks in individual patients.
In this study, Claude Gérard, Mickael Di-Luoffo, Léolo Gonay, and Frédéric Lemaigre identified a functional gene network involved in HCC and gastrointestinal cancers (see figure below). The network status negatively correlates with HCC vital prognosis and positively correlates with hyperproliferation of the tumor cells.
 |
|
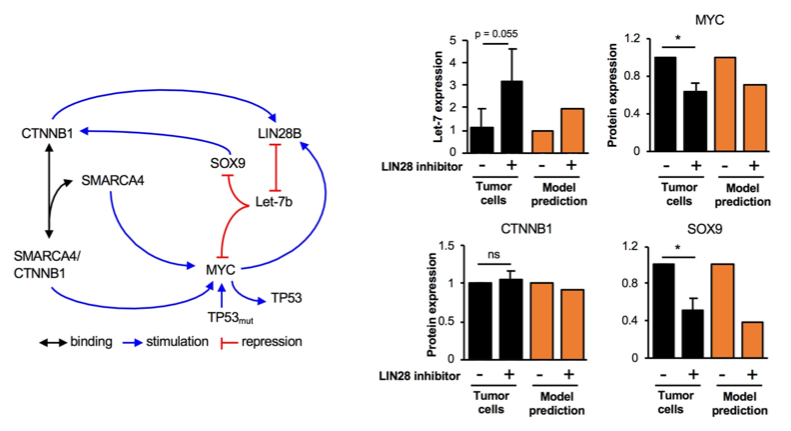
Gene network driving hepatocellular carcinoma. Left: structure of the network. CTNNB1 (beta-catenin), MYC, LIN28 are oncogenes in HCC, TP53 and Let-7 are tumor suppressors. Right: effect of a drug targeting LIN28 on other genes of the network, as measured in primary tumor cells (black) or as predicted by the mathematical model. |
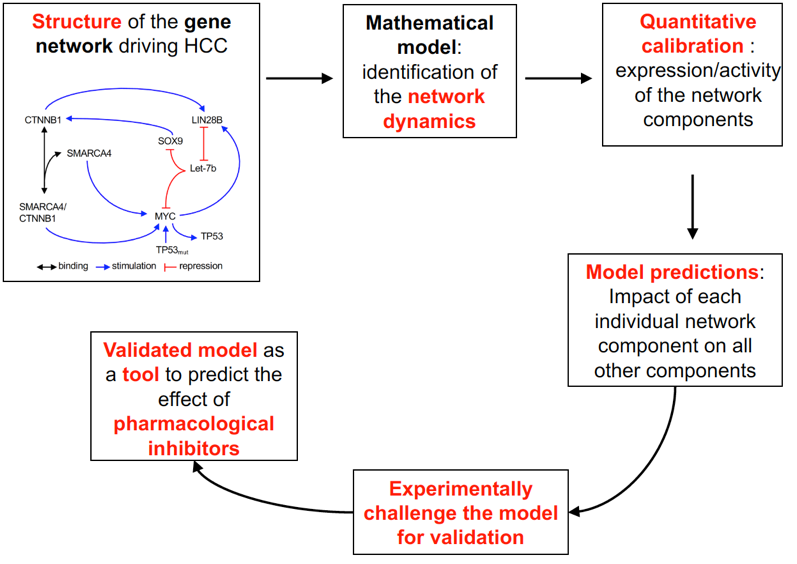
Claude Gérard, Mickael Di-Luoffo, Léolo Gonay, and Frédéric Lemaigre, with help of the teams of Jessica Zucman-Rossi (Centre des Cordeliers, Paris), Paul Monga (University of Pittsburgh), Emmanuel Hanert (UCLouvain, Louvain-la-Neuve), and Jens Marquardt (Johannes Gutenberg University, Mainz) developed a mathematical model of a gene network driving hepatocellular carcinoma. The model consists of a set of equations describing the interactions of the genes within the network, and the values integrated in the model (expression and/or activity of each network component) were measured experimentally in cultured cells and patient samples. Using the model we predicted how a therapeutic agent targeting one gene of the network affects the activity of all the genes belonging to the network. The methodological steps are illustrated in the figure below:
 |
Our approach also helps in characterising how the tumor will progress and so contributes to determine the prognosis of the disease in individual patients. The mathematical model is made available to the biomedical community via a web-based tool.
Hepatocellular carcinoma is driven by several such networks. We anticipate that identification and modeling of a set of dysregulated networks will contribute to provide a global picture of tumor-promoting gene function in liver cancer. Therefore, the study presents the concept of tools that help in the design of bespoke therapies for treating the disease.
Article describing this research
Gérard C, Di-Luoffo M, Gonay L, Caruso S, Couchy G, Loriot A, Castven D, Tao J, Konobrocka K, Cordi S, Monga SP, Hanert E, Marquardt JU, Zucman-Rossi J, Lemaigre FP.
Journal of Hepatology (2019) Apr 3. pii: S0168-8278(19)30195-3. doi: 10.1016/j.jhep.2019.03.024.


