We try to understand why tumor-infiltrating lymphocytes are unable to kill tumor cells, and to find clinical strategies to overcome this blockage. We have discovered that the dysfunction of human tumor-infiltrating lymphocytes can be due to the presence of galectin-3, a lectin abundant in tumors.
T cells can kill cancer cells
That our immune system could get rid of some cancers is a dream whose experimental bases go back to the 40s. Mice whose tumor had been surgically removed were able to eliminate upon injection cells from the same tumor but were not able to eliminate cells from another tumor. This showed the intervention of the immune system, which works with some specialized cells among which some white blood cells, called T lymphocytes, carry receptors that can recognize foreign molecular structures, named antigens. We have tens of millions of different T lymphocytes, each carrying a single type of receptor capable of recognizing a particular antigen. This antigen recognition by the T cell will lead to the proliferation and the activation of the lymphocyte. Some become cytolytic lymphocytes, capable firstly to burst the cell on which they recognize their antigen, and secondly to produce molecules which serve as messages for other cells. One of these messages is interferon-gamma.
The discovery of tumor antigens
T cells can cure viral infections because the cells infected by viruses carry viral antigens. Nobody knew, until twenty years ago, if there were specific antigens on human tumors and thus whether human T cells could distinguish tumor cells from normal cells. It is now clear that the majority of human cancers present antigens specific for cancer cells. The de Duve Institute has played a key role in demonstrating this concept because the first tumor antigens have been discovered here by the team of Thierry Boon. Pierre van der Bruggen and Catia Traversari were at the forefront. Dozens of these antigens recognized by cytolytic T lymphocytes are now known, and many remain to be discovered. But some of them already have characteristics that make them excellent candidates for the development of a therapy based on the activation of anti-tumor T lymphocytes. The presence of tumor antigens results from the expression of genes, e.g. MAGE, that are expressed in many tumors but not in normal tissue (Figure 1).
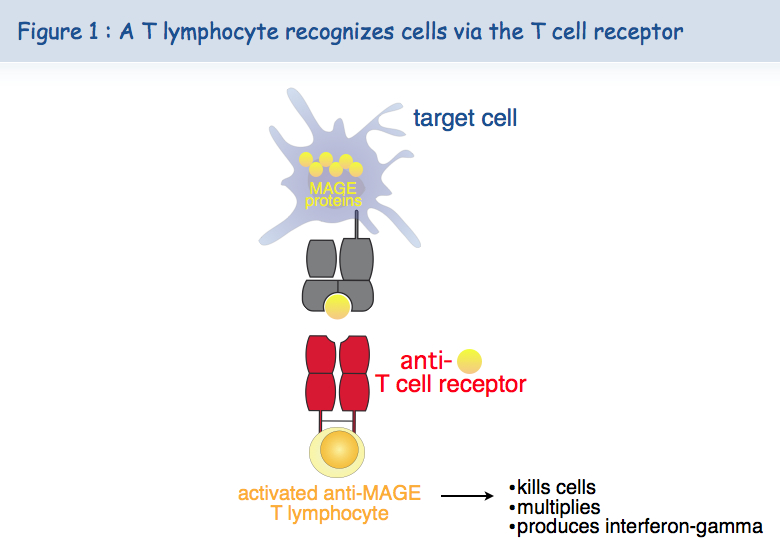
Further to this tumor specificity, a second feature of the MAGE antigens is their presence on many different cancers, allowing the use in many patients of the same therapeutic vaccine. We talk here about therapeutic vaccines versus prophylactic vaccines.
Anti-tumor therapeutic vaccination
The first therapeutic vaccination with MAGE antigens began in 1994 in patients with melanoma. Among vaccinated patients, 15% showed a significant regression of their cancer while the frequency of spontaneous regression in metastatic melanoma is estimated at less than 0.5%. These regressions, sometimes complete and durable, were obtained in the absence of any side effects, which seems to be a decisive advantage over conventional cancer treatments such as chemotherapy.
The key for the future will be to understand why therapeutic vaccination fails in most cases. Have we been unable to trigger a lymphocyte response in these patients? Is their tumor resistant to immune attack? And in each case, why?
T cells that are found in tumors are exhausted
At the beginning of our clinical program, we thought that the immune system of a cancer patient remained "asleep" in front of antigens expressed by the tumor, but that a vaccine containing the right ingredients could induce strong T cell responses that would destroy cancer cells.
Various observations made by the team of Pierre Coulie at the de Duve Institute and that of Thierry Boon led to propose an alternative scenario to explain the tumor regressions induced by vaccination. Patients with metastatic melanoma develop spontaneous immune responses against some tumor antigens expressed by their tumor. These anti-tumor lymphocytes are concentrated in metastases, but become unable to act effectively against tumor cells, presumably because of resistance mechanisms set up by the tumor. In some patients, a small number of T cells induced by the vaccine could migrate into the tumor and could, by an unknown mechanism, activate or reactivate other anti-tumor lymphocytes, allowing them to proliferate and destroy the cancer cells (Figure 2).

A new mechanism of exhaustion of lymphocytes
Analysis on lymphocytes grown in the laboratory
Several studies indicate that tumor-infiltrating lymphocytes are not working properly. This is known as T cell dysfunction. Researchers from Pierre van der Bruggen's team discovered a new mechanism of dysfunction of human T lymphocytes and found approaches to correct this dysfunction. They initially observed that most cytolytic T lymphocytes, after contact with antigen-presenting cells, lose for several days their capacity to kill antigen-presenting cells and to secrete interferon-gamma.
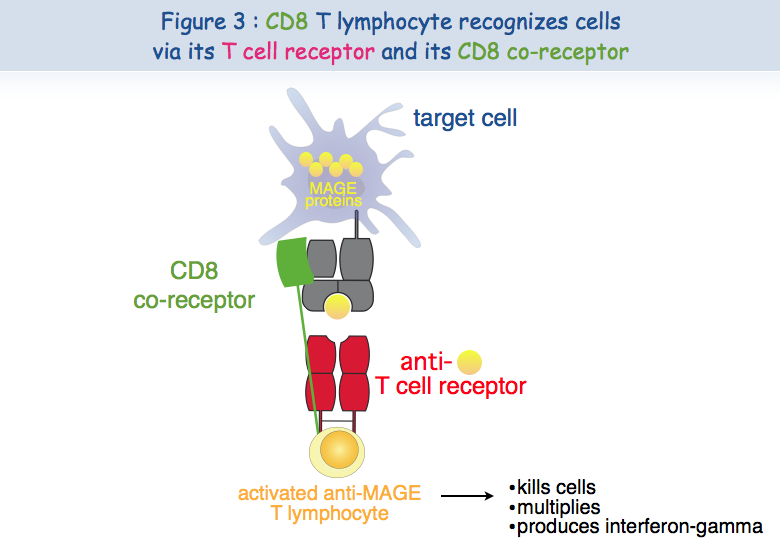
Different experimental approaches have been implemented to try to understand this dysfunction. A cytolytic T cell requires at least two receptors in order to recognize the antigen and be activated: TCR and CD8 (Figure 3).
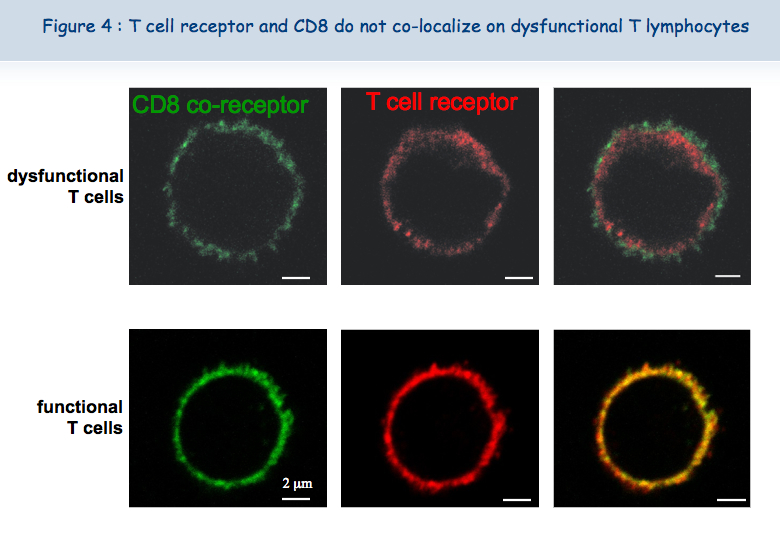
The new microscope recently acquired by the de Duve Institute and the skills of Pierre Courtoy and Patrick Van Der Smissen from the Cell Biology unit have shown that the two receptors are close to each other (co-localized) at the surface of functional lymphocytes, while these two receptors are not co- localized at the surface of dysfunctional lymphocytes (Figure 4).
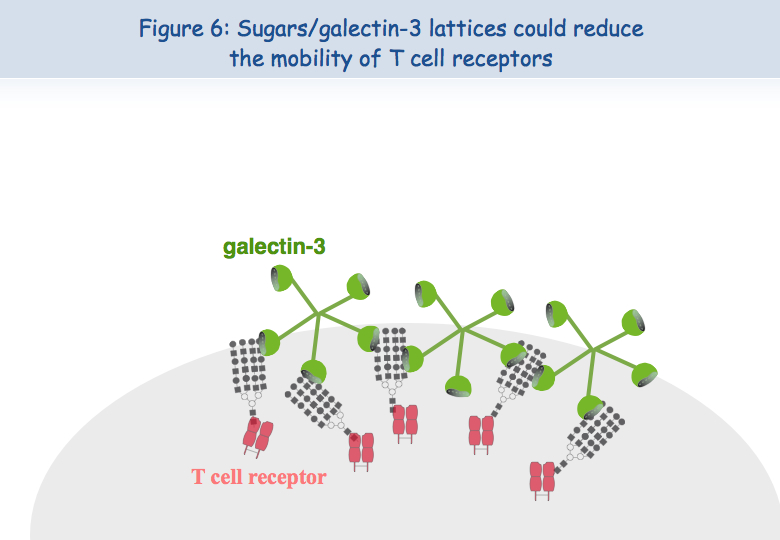
To explain this lack of co-localization, the researchers considered the possibility that these receptors are retained in different locations of the membrane. Regarding the TCR, it seems that when the lymphocytes are put in the presence of the antigen too frequently, the TCR loads "sugars". A molecule called galectin-3 then binds to these sugars and thus binds the TCR. TCR lose their mobility and cannot interact with the other receptor, CD8 (Figures 5, 6 & 7).

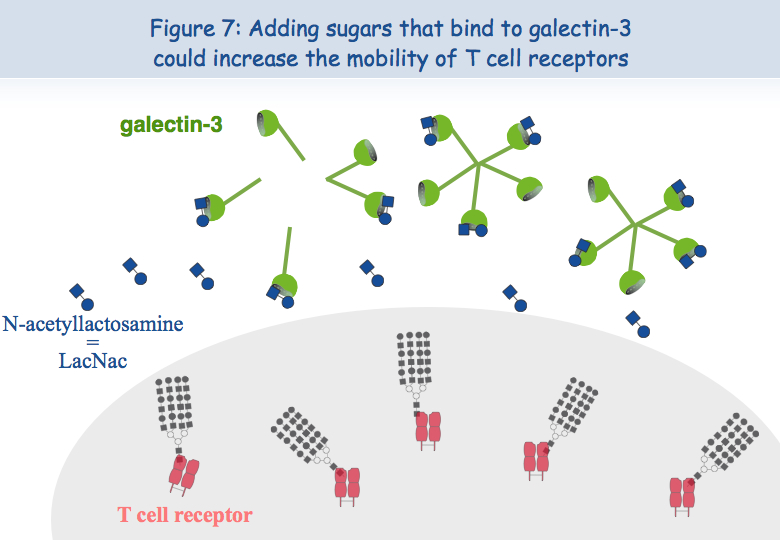
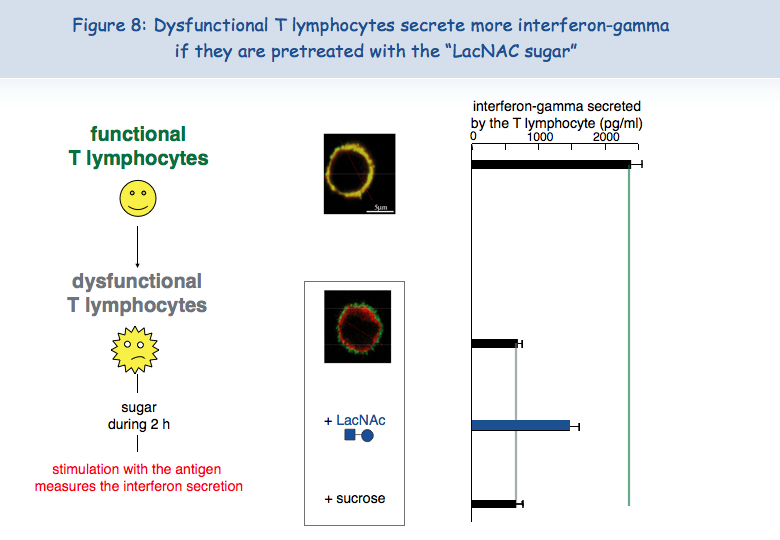
To test this hypothesis of a lack of mobility of the TCR at the surface of dysfunctional T cells, these were put in the presence of a sugar that binds to galectin-3, LacNAc. Two hours of treatment with LacNAc allowed the recovery of a large part of the TCR-CD8 co-localization. T cells treated with LacNAc also recovered their ability to secrete interferon-gamma following stimulation with the antigen (Figure 8).
Analysis of lymphocytes infiltrating the tumors of patients
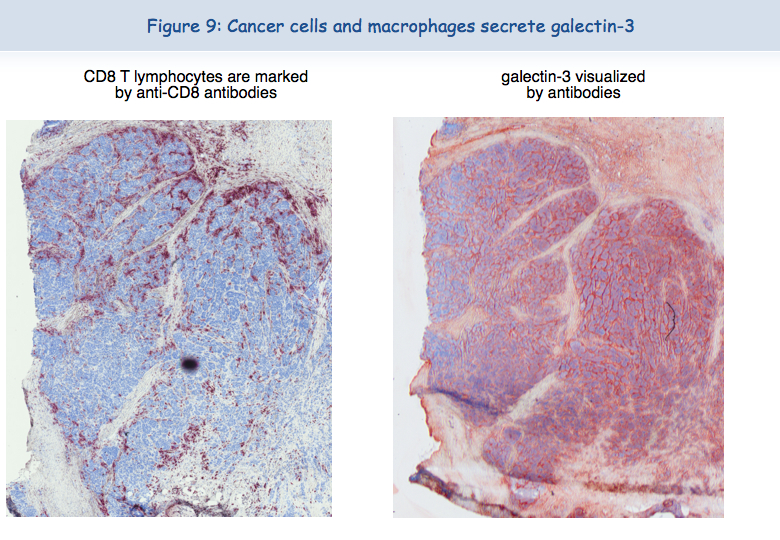
There are very often large amounts of galectin-3 in tumors (Figure 9).
It was tempting to think that, as in T cells cultured in the laboratory, the T cells that are found in tumors were dysfunctional due to the presence of galectin-3. With the collaboration of physicians at the Cliniques Saint-Luc (Brussels), we were able to isolate human T cells from tumors. At the surface of these T lymphocytes, TCR and CD8 were not co-localized. These lymphocytes were unable to secrete interferon-gamma following stimulation with tumor cells. These results clearly indicated that the dysfunction of tumor-infiltrating lymphocytes was correlated with a lack of TCR-CD8 co-localization.
Restoring functions of exhausted T cells
The following observation was the most important: it was possible to restore the functions of tumor-infiltrating lymphocytes. After overnight incubation in the presence of LacNAc (the sugar that binds to galectin-3), lymphocytes had recovered the TCR-CD8 co-localization and the ability to secrete interferon-gamma (Figure 10).

These results obtained ex vivo allow us to suggest that injecting tumors with LacNAc or other galectin antagonists could restore at least temporarily the functions of T cells and thus create favorable conditions for an effective anti-tumor response. Combined with therapeutic vaccination, treatment with this type of sugar may induce tumor regression in a larger number of patients. The team of Pierre van der Bruggen is currently testing different types of sugar that could be injected in cancer patients and, in vitro, the initial results are promising.
More information
Demotte N, Stroobant V, Courtoy PJ, Van Der Smissen P, Colau D, Luescher IF, Hivroz C, Nicaise J, Squifflet JL, Mourad M, Godelaine D, Boon T, van der Bruggen P. Restoring the association of the T cell receptor with CD8 reverses anergy in human tumor-infiltrating lymphocytes. Immunity, 2008, 28:414-24
Boon T, Coulie PG, Van den Eynde B, van der Bruggen P. Human T cell responses against melanoma. Annu. Rev. Immunol., 2006, 24:175-208.
The analysis of the T cell responses of melanoma patients vaccinated against tumor antigens has led to consider the possibility that the limiting factor for therapeutic success is not the intensity of the anti-vaccine response but the degree of anergy presented by intratumoral lymphocytes. We try to understand why tumor-infiltrating lymphocytes are unable to kill tumor cells, and to find strategies to overcome this blockage. We have discovered a new type of anergy of human tumor-infiltrating lymphocytes, due to the presence of galectin-3, a lectin abundant in tumors, as it is corrected by galectin antagonists. We are further analyzing the mechanisms by which galectin antagonists reverse the impaired function and we look for agents that correct this function. A clinical trial combining anti-tumoral therapeutic vaccination and injection of galectin antagonists was launched in 2013 in clinical centers close to the de Duve Institute.
Previous work in our group: Identification of tumor antigens recognized by T cells
In the 1970s it became clear that T lymphocytes, a subset of the white blood cells, were the major effectors of tumor rejection in mice. In the 1980s, human anti-tumor cytolytic T lymphocytes (CTL) were isolated in vitro from the blood lymphocytes of cancer patients, mainly those who had melanoma. Most of these CTL were specific, i.e. they did not kill non-tumor cells. This suggested that they target a marker, or antigen, which is expressed exclusively on tumor cells. We started to study the anti-tumor CTL response of a metastatic melanoma patient and contributed to the definition of several distinct tumor antigens recognized by autologous CTL. In the early 1990s, we identified the gene coding for one of these antigens, and defined the antigenic peptide. This was the first description of a gene, MAGE-A1, coding for a human tumor antigen recognized by T lymphocytes.
Genes such as those of the MAGE family are expressed in many tumors and in male germline cells, but are silent in normal tissues. They are therefore referred to as “cancer-germline genes”. They encode tumor specific antigens, which have been used in therapeutic vaccination trials of cancer patients. A large set of additional cancer-germline genes have now been identified by different approaches, including purely genetic approaches. As a result, a vast number of sequences are known that can code for tumor-specific shared antigens. The identification of a larger set of antigenic peptides, which are presented by HLA class I and class II molecules and recognized on tumors by T lymphocytes, could be important for therapeutic vaccination trials of cancer patients and serve as tools for a reliable monitoring of the immune response of vaccinated patients. To that purpose, we have used various approaches that we have loosely named “reverse immunology”, because they use gene sequences as starting point.
Human tumor antigens recognized by CD4+ or CD8+ T cells are being defined at a regular pace worldwide. Together with colleagues at the de Duve Institute, we read the new publications and incorporate the newly defined antigens in a database accessible at http://cancerimmunity.org/peptide
Further reading
van der Bruggen P, Zhang Y, Chaux P, Stroobant V, Panichelli C, Schultz ES, Chapiro J, Van den Eynde BJ, Brasseur F, Boon T. 2002. Tumor-specific shared antigenic peptides recognized by human T cells. Immunological Reviews, 188: 51-64
van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, Knuth A, Boon T. 1991. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science, 254: 1643-1647
A mechanism causing anergy of CD8 and CD4 T lymphocytes
The identification of specific tumor antigens recognized by T lymphocytes on human cancer cells has elicited numerous clinical trials involving vaccination of tumor-bearing cancer patients with defined tumor antigens. These treatments have shown a low clinical efficacy. Among metastatic melanoma patients, about 5% show a complete or partial clinical response following vaccination, whereas an additional 10% show some evidence of tumor regression without clear clinical benefit. We believe that progress depends on unraveling the different blockages for efficient tumor destruction.
The tumors of the patients about to receive the vaccine, already contain T cells directed against tumor antigens. Presumably these T cells are exhausted and this impaired function is maintained by immunosuppressive factors present in the tumor. The T cell response observed in some vaccinated patients reinforce an hypothesis proposed by Thierry Boon and Pierre Coulie: anti-vaccine CTL are not the effectors that kill the tumor cells but their arrival at the tumor site containing exhausted anti-tumor CTL, generates conditions allowing the reawakening of the exhausted CTL and/or activation of new anti-tumor CTL clones, some of them contributing directly to tumor destruction. Accordingly, the difference between the responding and the non-responding vaccinated patients is not the intensity of their direct T cell response to the vaccine but the intensity of the immunosuppression inside the tumor. It is therefore important to know which immunosuppressive mechanisms operate in human tumors.
Dysfunction of T lymphocytes can be corrected by targeting galectins
CD8 T cell clones
CTL clones can be maintained in culture by stimulation every 1-2 weeks with cells presenting the antigen, in the presence of growth factors and EBV-B transformed cells as feeder cells. The functional status of CTL clones can be checked regularly by testing their capacity to lyse target cells expressing the relevant antigen and to produce cytokines upon antigenic stimulation, e.g. IFN-γ. Fluorescent HLA-peptide complexes –multivalent complexes of HLA molecules folded in the presence of the antigenic peptide and coupled to a fluorochrome– can be used to visualize antigen-specific CTL bearing the appropriate TCR (90). We observed that, compared with resting CTL collected 14 days after the last stimulation, recently activated CTL collected four days after stimulation have lost their capacity to bind HLA-tetramer complexes. They also secrete lower levels of cytokines upon a further antigenic stimulation (91). The decreased tetramer labeling and function was not due to a reduced surface expression of either the TCR or the CD8 co-receptor, which are both essential for tetramer labeling and activation of T lymphocytes. We decided therefore to examine by confocal microscopy the surface distribution of TCR and CD8 molecules on resting and recently activated T cells. TCR and CD8 molecules appeared to be co-localized on resting CTL, whereas TCR were segregated from CD8 molecules at the surface of recently activated CTL. These results were confirmed by fluorescence resonance energy transfer (FRET), where interactions between two proteins can be estimated at a resolution of 10 nm.
Our hypothesis to explain the separation of TCR and the CD8 molecules at the cell surface of recently activated CTL is inspired by work from the group of Dennis and Demetriou (92, 93): N-glycosylated TCR molecules are clustered by extracellular galectin-3 and form glycoprotein-galectin lattices, which decrease the lateral mobility of TCR. In agreement with this hypothesis we observed that recently activated CTL, which were treated for 2 h with mM concentrations of galectin ligand N-acetyllactosamine (LacNAc), secreted more IFN-γ upon antigenic stimulation. Moreover this short LacNAc treatment restored TCR-CD8 co-localization, as measured by FRET.
Tumor-infiltrating lymphocytes
We collected a large number of ascites samples from patients with various tumors, in particular ovarian and pancreatic carcinoma. We also collected samples of solid tumors, mostly melanoma. CD8 T lymphocytes were isolated from these samples and tested ex vivo ‑without prior in vitro expansion– for their capacity to secrete IFN-γ upon non-specific stimulation, using beads coated with anti-CD3 and anti-CD28 antibodies. CD8 TIL from most of the samples secreted low levels of IFN-γ, in contrast with CD8 blood lymphocytes. Secretion of other cytokines by TIL was also low, e.g. IL-2 and TNF-α. These results are in line with the very few studies that have demonstrated dysfunction of human TIL (94-98).
Treating CD8 TIL for a few hours with mM concentrations of LacNAc increased by at least three times the secretion of IFN-γ, IL-2 and TNF-α (99). This holds true for 80% of the samples tested so far. LacNAc-treated CD4 TIL were also able to secrete high amounts of IFN-γ upon stimulation. The cytotoxicity of CD8 TIL was tested in a redirected killing assay, where the targets were mouse cells decorated with anti-CD3 antibodies. The cytotoxicity of CD8 TIL was minimal compared to the cytotoxicity of blood CD8 T lymphocytes, but increased greatly after an overnight LacNAc treatment.
GCS-100, a modified citrus pectin, and GM-CT-01, a galactomannan extracted from guar beans, are polysaccharides that could bind to galectins and have already been injected in humans (100-102). A short treatment of CD8 TIL with mM concentrations of GCS-100 boosted their cytotoxicity to an efficiency level similar to LacNAc treatment. It also boosted the secretion of cytokines by either CD8 or CD4 TIL (99). Experiments with GM-CT-01 are ongoing and the results are very encouraging (manuscript in preparation).
LacNAc, GCS-100, and GM-CT-01 can interact with several galectins and therefore it is difficult to attribute the effect on TIL functions of these sugars to one galectin in particular. To understand which galectins were implicated, we searched for the presence of different extracellular galectins at the TIL surface. We detected both galectin-1 and galectin-3, but failed to detect galectin-8, -9 or MGL, a lectin implicated in the regulation of T-cell function (103). Treating TIL with anti-galectin-3 antibody B2C10 boosted IFN-γ secretion upon stimulation to levels similar to LacNAc or GCS-100 treatments (104). Because B2C10 was unable to detach galectin-1 while boosting TIL function, we concluded that detaching galectin-3 from TIL is sufficient to restore function, while not excluding a contribution of other galectins. We have so far failed to identify an anti-galectin-1 antibody able to detach galectin-1 from cells and are therefore unable to examine if galectin-1 also plays a role in TIL dysfunction.
How do galectins influence the distribution of T cell surface molecules?
Galectin-3 is an abundant lectin in many solid tumors and carcinomatous ascites. It can thus bind to surface glycoproteins of TIL. Glycoproteins often bear multiple copies of the sugar moieties that are recognized by galectins. The multivalent nature of galectin-glycan interactions results in high avidity in the range of 106 M−1, and allows the formation of galectin-glycoprotein lattices (reviewed elsewhere (105)). Similarly, lattices on TIL, formed by glycosylated surface receptors and extracellular galectin-3, would reduce the mobility of the former molecules, a fact that could explain the impaired function of TIL. The release of galectin-3 by soluble galectin ligands would restore the mobility of glycosylated surface receptors and boost IFN-γ secretion by TIL. Anti-galectin-3 antibody B2C10 should have a similar effect: this antibody binds to the N-terminal region of the galectin-3 and its rigid structure could prevent association of galectin-3 monomers mediated by the N-terminal region, thereby affecting the oligomerization of the lectin. Antibody B2C10 was shown to inhibit erythrocyte agglutination mediated by galectin-3 oligomerization (104).
The presence of galectin-glycoprotein lattices is in agreement with our observations with CTL clones and TIL. TCR and CD8 molecules are not co-localized on dysfunctional T cells and treating dysfunctional T cells with either an anti-galectin-3 antibody or galectin ligands detached galectin-3 from the T cell surface and restored the TCR-CD8 co-localization estimated by FRET (91, 99). Considering that HLA-peptide tetramer binding requires TCR and CD8 cooperation, the disorganization of galectin-glycoproteins lattices in the presence of galectin ligands could explain the recovery of HLA-peptide tetramer binding on dysfunctional CTL clones that were treated with LacNAc (91).
A number of publications suggest that several human T cell surface glycoproteins can be linked together by galectins and form lattices. Human TCR α-chain, in contrast to the β-chain, has been shown to harbor complex N-glycans, the major natural ligands for galectin-3 (106, 107). The removal of an N-glycosylation site from a human TCR α-chain was reported to result in increased avidity of T cells grafted with the gene encoding the modified TCR (108). Galectin-3 was reported to bind to CD45, CD29, CD43, and CD71 (109), and galectin-1 to CD45, CD7 and CD43 (110). Galectin-1/CD7 ligation was shown to induce apoptosis of CD4 T lymphocytes (111). It has also been shown by immunoprecipitation that galectin-3 binds to CD45 and that galectin-3 influences the CD45 partition to microdomains containing the TCR (112). Interestingly, CD45-TCR proximity is known to negatively regulate the TCR signaling cascade (113).
Glycosylation changes at the T cell surface
To explain that galectin-3, alone or together with galectin-1, inhibits functions of recently activated T cells, we surmised that the recently activated T cells, compared to resting T cells, harbor a set of glycans that are either more numerous or better ligands for galectin-3. We stimulated resting CTL in the presence of swainsonine that inhibits α-mannosidase II involved in the N-glycosylation pathway. Compared to untreated cells, cells stimulated in the presence of swainsonine showed on day 4 a better TCR-CD8 co-localization and a higher ability to release IFN-γ upon antigenic stimulation (91).
We also characterized global changes in surface N- and O-glycans on two human CTL clones that were collected either in a resting state, 14 days after antigenic stimulation, or in a recently activated state, four days after antigenic stimulation. Applying ultra-sensitive MALDI-TOF-MS, combined with various glycosidase digestions and GC-MS linkage analyses, we made two novel observations.
Firstly, the N-glycome of recently activated cells versus resting cells consists of longer LacNAc chains, of which a portion contains more than four LacNAc moieties (poly-LacNAc). Secondly, it contains more multi-antennary N-glycans (114). Interestingly, our results showed that the above poly-LacNAc chains appeared to be equally distributed on all available N-glycan branches and not selectively enriched on a specific branch. In contrast, murine T cells are poor in tri- and tetra-antennary poly-LacNAc glycans (115). This difference could potentially explain the crucial role in mice of N-acetylglucosaminyltransferase V (Mgat5), an enzyme essential for the generation of complex tetra-antennary N-glycan, as T cells from mice lacking Mgat5, compared to their wildtype counterparts, are more sensitive to activation and have reduced poly-LacNAc motives (92). The glycome modifications observed on human CTL clones upon activation are expected to increase the number of galectin-3 natural ligands, but also the number of galectin-1 ligands, and favor lattices that could reduce the mobility of surface glycoproteins, as the affinity of both galectins was reported to be much higher when repeated LacNAc units are present (116).
We also observed that recently activated CTL clones exhibited a lower abundance of terminal a2,6-linked NeuAc residues than resting CTL (114). Galectin-1 binds to terminal LacNAc units, even if they are decorated with a2,3-linked NeuAc. On the contrary, galectin-1 binding is blocked by the presence of terminal a2,6-linked NeuAc (88, 110, 111, 117, 118). Our glycome analyses suggest therefore that more galectin-1 natural ligands are presented on recently activated CTL versus resting CTL.
All together, the results of our glycome analyses of CTL clones combined with the fact that functions of both CTL clones and TIL can be boosted by galectin ligands, support our working hypothesis: TIL are in permanent contact with tumor cells and have been stimulated by antigen recently. The resulting activation of T cells modifies the expression profiles of enzymes of the N-glycosylation pathway, increasing the expression of N-glycans at the T cell surface. Considering the high abundance of extracellular galectins in tumors, secreted by tumor cells and macrophages, this could favor the formation of galectin-glycoprotein lattices and therefore the dysfunction of some TIL.
Towards a clinical trial combining vaccination and galectin-binding polysaccharides
As described above, treating TIL with modified citrus pectin GCS-100 improved their ability to secrete IFN-γ upon stimulation. We decided therefore to test the therapeutic effect of GCS-100 in tumor-bearing mice. We injected 40 mice subcutaneously in the flank with 2x106 P815 mastocytoma cells. On day 4, half of the animals were vaccinated with an adenovirus encoding P815 tumor antigen P1A (5). Therapeutic vaccination has thus far proven ineffective at inducing tumor rejection in tumor-bearing mice (Catherine Uyttenhove & Guy Warnier, personal communication). On day 10, treatments with either PBS or GCS-100 were initiated three times a week. Three weeks later, the tumor had become undetectable in six out of the ten vaccinated mice treated with GCS-100, of which five were still alive after another three months. Control mice that received only the vaccine died. In non-vaccinated mice, the polysaccharide had no visible effect by itself. These results suggest that a combination of galectin-3 ligands and therapeutic vaccination may induce more effective tumor regression in cancer patients than vaccination alone. Setting up a clinical trial combining anti-tumoral vaccination and GCS-100 was impossible because the pharmaceutical company that produced the GCS-100 declared bankruptcy.
GM-CT-01, a galactomannan derived from guar gum, has been shown to bind to galectin-1 (119), and to increase the anti-tumor activity of chemotherapy drug 5-fluorouracil. It was previously injected in patients with solid tumors without major side effects (120). We have treated TIL samples with GM-CT-01 and the first results are encouraging. We therefore will launch a Phase I/II clinical trial combining peptide vaccination associated with intravenous injections of GM-CT-01 in patients with advanced melanoma (Fig.1). Patients will receive sequential vaccinations with one or two peptides, MAGE-3.A1 and NA17.A2, matching the tumor antigens expressed by their tumor. Their formulation and schedule of vaccination (timing, dose, route of administration) will be similar to previous trials with these peptides (15, 20, 21). GM-CT-01 will be administered systemically by repeated intravenous infusions, in order to ensure a prolonged effect on tumor-associated lymphocytes. The treatment dose matches the total cumulative dose in previous GM-CT-01 treatment schedules (120). For selected patients with cutaneous or subcutaneous metastases, in addition to systemic GM-CT-01, small amounts of this drug will be injected close to metastases, to increase local concentration of the drug.
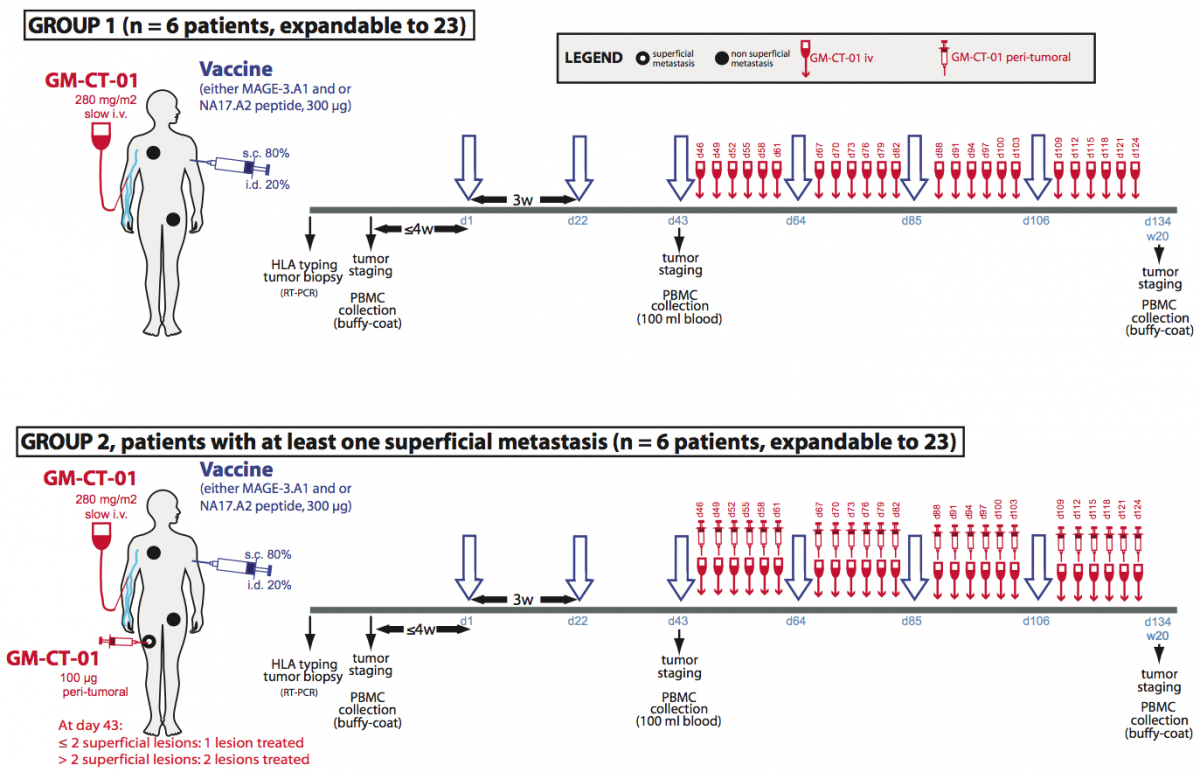
Figure 1. Treatment plan of the clinical trial involving GM-CT-01.
Treatment allocation: Patients will be divided in two treatment arms. Both will run in parallel. Patients with at least one measurable lesion will be assigned to group 1 and will receive the following treatment: peptide vaccinations and systemic GM-CT-01 injections. Patients with at least one measurable and at least one superficial metastasis will be assigned in priority to group 2 and will receive the following treatment: peptide vaccinations, systemic GM-CT-01 administrations and peri-tumoral administration of GM-CT-01 in one or two superficial metastases.
Human tumor-infiltrating T lymphocytes show impaired IFN-γ secretion
Both human CD8 and CD4 tumor-infiltrating T lymphocytes (TIL) were isolated from tumor ascites or solid tumors and compared with T lymphocytes from blood donors. TIL secrete low levels of INF-γ and other cytokines upon non-specific stimulation with anti-CD3 and anti-CD28 antibodies. TCR were observed to be distant from the co-receptors on the cell surface of TIL, either CD8 or CD4, whereas TCR and the co-receptors co-localized on blood T lymphocytes.

TCR and CD8 do not co-localize on CD8 T cells with impaired functions (in collaboration with P. Vandersmissen and P. Courtoy, PICT/SSS)
Reversing the anergy of tumor-infiltrating T lymphocytes with galectin antagonists
We have attributed the decreased IFN-γ secretion to a reduced mobility of T cell receptors upon trapping in a lattice of glycoproteins clustered by extracellular galectin-3. Indeed, we have shown that treatment of TIL with N-acetyllactosamine (LacNAc), a galectin antagonist, restored this secretion.

Treatment of tumor-infitrating lymphocytes with a galectin antagonist reverses
Our working hypothesis is that TIL have been stimulated by antigen chronically, and that the resulting activation of T cells could modify the expression of enzymes of the N-glycosylation pathway, as shown for murine T cells. The chronically activated TIL, compared to resting T cells, could thus express surface glycoproteins decorated with a set of glycans that are either more numerous or better antagonists for galectin-3. Galectin-3 is an abundant lectin in many solid tumors and carcinomatous ascites, and can thus bind to surface glycoproteins of TIL and form lattices that would thereby reduce TCR mobility. This could explain the impaired function of TIL. The release of galectin-3 by soluble competitor antagonists would restore TCR mobility and boost IFN-γ secretion by TIL. We recently strengthened this hypothesis by showing that both CD4 and CD8 TIL that were treated with an anti-galectin-3 antibody, which could disorganize lattice formation, had an increased IFN-γ secretion compared to untreated cells.
Towards a clinical trial combining vaccination and galectin-binding polysaccharides
Galectin competitor antagonists, e.g. disaccharides LacNAc, are rapidly eliminated in urine, preventing their use in vivo. We recently found that a plant-derived polysaccharide, currently in clinical development, detached galectin-3 from TIL and boosted their IFN-γ secretion. Importantly, we observed that not only CD8+ TIL but also CD4+ TIL that were treated with this polysaccharide secreted more IFN-γ upon ex vivo re-stimulation. In tumor-bearing mice vaccinated with a tumor antigen, injections of this polysaccharide led to tumor rejection in half of the mice, whereas all control mice died. In non-vaccinated mice, the polysaccharide had no effect by itself. These results suggest that a combination of galectin-3 antagonists and therapeutic vaccination may induce more tumor regressions in cancer patients than vaccination alone. Translation of these results to the clinic was unfortunately impossible because the company producing this polysaccharide got bankrupted. We recently identified another plant-derived polysaccharide that binds to galectins and was already used in combination with chemotherapy in phase II clinical trials in colorectal cancer patients. This compound was as effective as LacNAc in boosting the secretion of IFN-γ by treated TIL. A clinical trial with this new compound, in combination with anti-tumoral vaccination was launched in 2013 in clinical centers close to the de Duve Institute. We are currently trying to understand the very early activation events that are defective in TIL.
Further reading
Antonopoulos A, Demotte N, Stroobant V, Haslam SM, van der Bruggen P, Dell A. 2012. Loss of effector function of human cytolytic T lymphocytes is accompanied by major alterations in N- and O-glycosylation. Journal of Biological Chemistry, 287: 11240-11251
Demotte N, Wieërs G, Van Der Smissen P, Moser M, Schmidt CW, Thielemans K, Squifflet J-L, Weynand B, Carrasco J, Lurquin C, Courtoy PJ, van der Bruggen P. 2010. A galectin-3 ligand corrects the impaired function of human CD4 and CD8 tumor-infiltrating lymphocytes and favors tumor rejection in mice. Cancer Research, 70: 7476-7488
Demotte N, Stroobant V, Courtoy PJ, Van der Smissen P, Colau D, Luescher IF, Hivroz C, Nicaise J, Squifflet J-L, Mourad M, Godelaine D, Boon T, van der Bruggen P. 2008. Restoring the association of the T cell receptor with CD8 reverses anergy in human tumor-infiltrating lymphocytes. Immunity, 28: 414-424
Demotte N, Colau D, Ottaviani S, Godelaine D, Van Pel A, Boon T, van der Bruggen P. 2002. A reversible functional defect of CD8+ T lymphocytes involving loss of tetramer labeling. European Journal of Immunology, 32: 1688-1697
Petit AE, Demotte N, Scheid B, Wildmann C, Bigirimana R, Gordon-Alonso M, Carrasco J, Valitutti S, Godelaine D, van der Bruggen P.
Nat Commun. 2016; 7:12242.
Demotte N, Bigirimana R, Wieërs G, Stroobant V, Squifflet JL, Carrasco J, Thielemans K, Baurain JF, Van Der Smissen P, Courtoy PJ, van der Bruggen P.
Clin Cancer Res. 2014; 20(7):1823-33.
Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T.
Nat Rev Cancer. 2014; 14(2):135-46.
Demotte N, Wieërs G, Van Der Smissen P, Moser M, Schmidt C, Thielemans K, Squifflet JL, Weynand B, Carrasco J, Lurquin C, Courtoy PJ, van der Bruggen P.
Cancer Res. 2010; 70(19):7476-88.
François V, Ottaviani S, Renkvist N, Stockis J, Schuler G, Thielemans K, Colau D, Marchand M, Boon T, Lucas S, van der Bruggen P.
Cancer Res. 2009; 69(10):4335-45.
Demotte N, Stroobant V, Courtoy PJ, Van Der Smissen P, Colau D, Luescher IF, Hivroz C, Nicaise J, Squifflet JL, Mourad M, Godelaine D, Boon T, van der Bruggen P.
Immunity. 2008; 28(3):414-24.
Annu Rev Immunol. 2006; 24:175-208. Review.
Zhang Y, Sun Z, Nicolay H, Meyer RG, Renkvist N, Stroobant V, Corthals J, Carrasco J, Eggermont AM, Marchand M, Thielemans K, Wölfel T, Boon T, van der Bruggen P.
J Immunol. 2005; 174(4):2404-11.
Demotte N, Colau D, Ottaviani S, Godelaine D, Van Pel A, Boon T, van der Bruggen P.
Eur J Immunol. 2002; 32(6):1688-97.
Schultz ES, Chapiro J, Lurquin C, Claverol S, Burlet-Schiltz O, Warnier G, Russo V, Morel S, Lévy F, Boon T, Van den Eynde BJ, van der Bruggen P.
J Exp Med. 2002; 195(4):391-9.
Schultz ES, Lethé B, Cambiaso CL, Van Snick J, Chaux P, Corthals J, Heirman C, Thielemans K, Boon T, van der Bruggen P.
Cancer Res. 2000; 60(22):6272-5.
Chaux P, Luiten R, Demotte N, Vantomme V, Stroobant V, Traversari C, Russo V, Schultz E, Cornelis GR, Boon T, van der Bruggen P.
J Immunol. 1999; 163(5):2928-36.
Chaux P, Vantomme V, Stroobant V, Thielemans K, Corthals J, Luiten R, Eggermont AM, Boon T, van der Bruggen P.
J Exp Med. 1999; 189(5):767-78.
Mandruzzato S, Brasseur F, Andry G, Boon T, van der Bruggen P.
J Exp Med. 1997; 186(5):785-93.
Boël P, Wildmann C, Sensi ML, Brasseur R, Renauld JC, Coulie P, Boon T, van der Bruggen P.
Immunity. 1995; 2(2):167-75.
van der Bruggen P, Bastin J, Gajewski T, Coulie PG, Boël P, De Smet C, Traversari C, Townsend A, Boon T.
Eur J Immunol. 1994; 24(12):3038-43.
van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, Knuth A, Boon T.
Science. 1991; 254(5038):1643-7.

 Download
Download DYSFUNCTION OF T LYMPHOCYTES IN TUMORS
