Suivez-nous 

![]()
L'objectif principal de notre recherche est la découverte des variations génétiques qui entraînent certaines maladies humaines, et de comprendre comment et pourquoi elles le font. Nous étudions une variété de troubles du développement des systèmes cardio-vasculaire et squelettique, ainsi que certains cancers. Les mécanismes moléculaires d'un grand nombre d'entre eux ne sont pas encore connus, et les traitements actuels sont donc basés sur un large spectre, visant à soulager les symptômes. L'identification des causes primaires ainsi que des facteurs de modulation faciliterait grandement le développement de traitements plus efficaces, et avec moins d'effets secondaires. Pour atteindre cet objectif, nous utilisons des techniques génétiques de pointe sur des échantillons de patients pour la découverte de gènes, suivie par la génération de modèles cellulaires et animaux qui nous permettent de disséquer davantage les processus de la maladie et l'essai de nouvelles thérapies. Toutes nos recherches commencent avec des échantillons provenant de patients, et nous tentons de faire le tour du cercle: du lit au bench, pour revenir au lit, en fournissant aux patients des améliorations fondées sur les connaissances viables et tangibles sur leur santé.
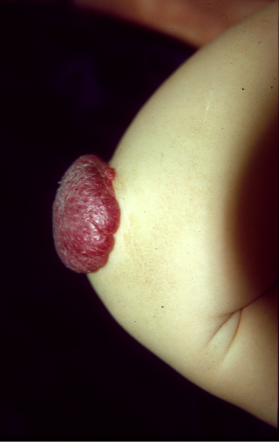
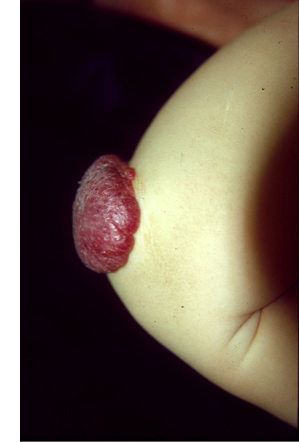 Les systèmes vasculaires sanguins et lymphatiques sont cruciaux pour le développement, la survie et le fonctionnement de l'organisme humain. Dans un processus complexe qui repose sur une large gamme de molécules pour fournir les bons signaux aux bons moments et aux bons endroits, un réseau bien organisé d'artères, de veines, de capillaires et de vaisseaux lymphatiques est formé. Les déséquilibres qui se produisent pendant ce processus résultent en une formation de défauts localisés, collectivement appelés anomalies vasculaires, qui sont classés selon le type de vaisseau(x) qu'ils affectent. L'International Society for the Study of Vascular Anomalies (ISSVA) a récemment détaillé leur classification. Ces déséquilibres peuvent être causés par des mutations dans les gènes codant pour des protéines impliquées dans le développement vasculaire. Nous avons identifié des mutations pour de nombreux types d'anomalies vasculaires différents, entre autres les malformations veineuses et lymphatiques, le lymphoedème, et les malformations capillaires combinées avec des malformations artério-veineuses. En général, les formes familiales de ces anomalies sont très rares, et sont causées par des mutations germinales. Leurs homologues sporadiques sont beaucoup plus fréquentes, et généralement causées par des mutations somatiques, à savoir, des mutations qui ne sont présentes que dans les zones touchées (malformées) et, par conséquent, non transmis d'une génération à l'autre. Les mutations géniques qui causent des anomalies vasculaires affectent presque toujours les cellules endothéliales qui forment la paroi interne des vaisseaux sanguins, et nous avons fait des progrès considérables dans la compréhension des changements qu'ils provoquent dans ces cellules, et comment ils peuvent être corrigés. Les essais cliniques sur base de ces données sont en cours au Centre des Anomalies Vasculaires.
Les systèmes vasculaires sanguins et lymphatiques sont cruciaux pour le développement, la survie et le fonctionnement de l'organisme humain. Dans un processus complexe qui repose sur une large gamme de molécules pour fournir les bons signaux aux bons moments et aux bons endroits, un réseau bien organisé d'artères, de veines, de capillaires et de vaisseaux lymphatiques est formé. Les déséquilibres qui se produisent pendant ce processus résultent en une formation de défauts localisés, collectivement appelés anomalies vasculaires, qui sont classés selon le type de vaisseau(x) qu'ils affectent. L'International Society for the Study of Vascular Anomalies (ISSVA) a récemment détaillé leur classification. Ces déséquilibres peuvent être causés par des mutations dans les gènes codant pour des protéines impliquées dans le développement vasculaire. Nous avons identifié des mutations pour de nombreux types d'anomalies vasculaires différents, entre autres les malformations veineuses et lymphatiques, le lymphoedème, et les malformations capillaires combinées avec des malformations artério-veineuses. En général, les formes familiales de ces anomalies sont très rares, et sont causées par des mutations germinales. Leurs homologues sporadiques sont beaucoup plus fréquentes, et généralement causées par des mutations somatiques, à savoir, des mutations qui ne sont présentes que dans les zones touchées (malformées) et, par conséquent, non transmis d'une génération à l'autre. Les mutations géniques qui causent des anomalies vasculaires affectent presque toujours les cellules endothéliales qui forment la paroi interne des vaisseaux sanguins, et nous avons fait des progrès considérables dans la compréhension des changements qu'ils provoquent dans ces cellules, et comment ils peuvent être corrigés. Les essais cliniques sur base de ces données sont en cours au Centre des Anomalies Vasculaires.
Liens utiles
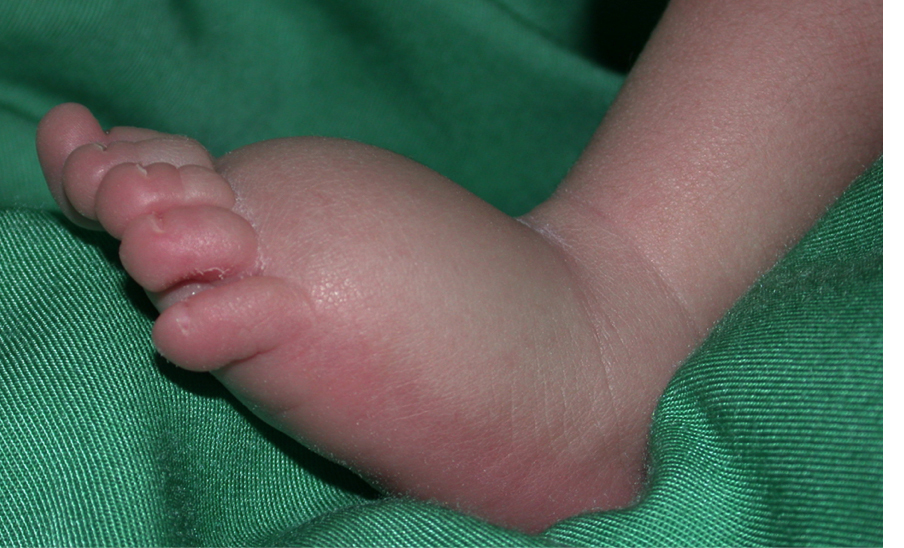
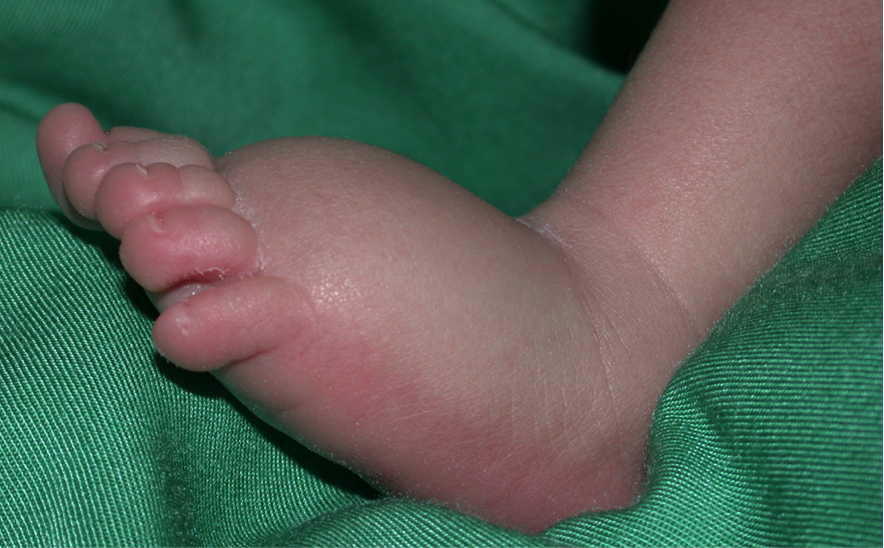
Le lymphoedème est une maladie chronique très invalidante résultant d'un développement et/ou d'un fonctionnement anormal du système lymphatique. La lymphe n'est pas évacuée des tissus interstitiels, mais s'accumule, le plus souvent dans les extrémités inférieures, ce qui provoque dilatation et fibrose, et prédispose aux infections secondaires. Le fonctionnement de la partie du corps touchée est souvent compromis. En Europe, plus d'un million de personnes sont touchées. Les thérapies sont limitées à du drainage lymphatique manuel répété, et à l'utilisation de moyens compressifs. Dans certains cas, la chirurgie peut être utile, mais aucun traitement curatif n'existe.
Les lymphœdèmes sont divisés en deux catégories : primaire (cause inconnue) et secondaire (cause environnementale connue, par exemple après une infection ou une thérapie du cancer). Le lymphoedème primaire est parfois hérité de génération en génération, de manière similaire aux anomalies vasculaires. En étudiant ces familles, nous avons découvert plusieurs gènes responsables du lymphoedème primaire, et démontré que les mutations peuvent être dominantes, récessives ou même de novo. Certaines provoquent un dysfonctionnement du système lymphatique foetal beaucoup plus généralisé (hydrops fetalis), ou des syndromes, nécessitant de les considérer lors d'un diagnostic. À ce jour, 28 gènes sont déjà connus pour causer un lymphoedème primaire et/ou prédisposer à la forme secondaire, mais ceux-ci ne représentent que moins d'un tiers des échantillons, chaque gène expliquant un faible pourcentage des cas.
Liens utiles
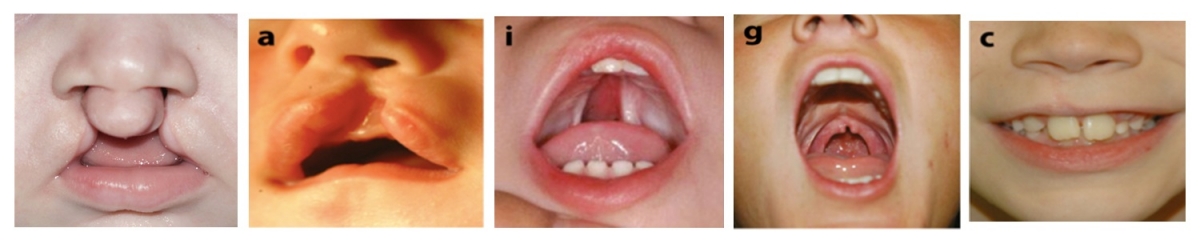
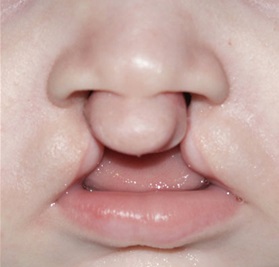
Les fentes labiales et/ou palatines (CL/P) constituent les malformations craniofaciales congénitales les plus fréquentes chez l’homme. Comme pour d'autres maladies humaines telles que l'hypertension artérielle et le diabète, une origine multifactorielle aux CL/P est évoquée, correspondant à une interaction complexe entre facteurs environnementaux et facteurs génétiques. Dans certaines formes familiales de CL/P, certaines variations génétiques peuvent expliquer la pathologie. Dans d’autres cas, certaines variations semblent prédisposer aux CL/P. Ainsi, la complexité des étiologies des CL/P rend difficile l’identification des gènes impliqués dans la pathologie.
Les formes clairement héréditaires de CL/P isolées seront étudiées, par l’analyse du génome de plusieurs personnes atteintes et de la coségregation de variations de gène d’intérêt au sein des familles. Ces gènes d’intérêt seront ensuite étudiés au sein d’une population de CL/P « sporadiques », à la recherche de variations retrouvées de façon plus significative que dans la population générale, et qui pourraient expliquer une prédisposition voire un effet protecteur.
Les CL/P peuvent également exister dans des formes syndromiques, c’est-à-dire coexister chez une personne avec d'autres signes cliniques. Nous avons découvert que les gènes responsables de certaines formes syndromiques contribuent également aux CL/P isolées lorsqu'ils sont moins « sévèrement » mutés. Cela constitue une piste supplémentaire pour essayer d’identifier des gènes candidats impliqués dans la maladie.
En collaboration avec d'autres groupes de recherche, nous cherchons à identifier les mutations "driver" et modificatrices (par exemple, des mutations qui déclenchent la maladie par rapport à celles qui affectent la progression) dans une variété de cancers: tumeurs neuroendocrines et cérébrales, cancer du sein et cancers hématologiques. De plus, nous cherchons à trouver des marqueurs qui aideront dans le diagnostic et le pronostic. Ceux-ci comprennent des changements génétiques (pour le diagnostic ou la classification «moléculaire»), ainsi que des protéines détectées dans la circulation ou dans des échantillons de tissus prélevés par biopsie.
Avec le Prof. D. MANICOURT
Nous essayons d'identifier le(s) gène(s) responsable(s) du syndrome d'Ehlers-Danlos (EDS), de type hypermobilité, une condition méconnue et pourtant relativement fréquente. Le syndrome n'a pas de traitement et aucune découverte de collagène biochimique distinctif. En raison de leur gamme excessive de mouvement, les articulations sont sujettes à la dislocation et aux subluxations articulaires. Le syndrome est une cause fréquente d'arthrose sévère, précoce et multifocale. D'autres manifestations cliniques incluent douleur chronique, ecchymoses, troubles intestinaux fonctionnels, dysautonomie et dilatation aortique.

 Le domaine de la génétique humaine est actuellement révolutionné par le séquençage à haut débit ("Next Generation Sequencing" ou "NGS"). D'énormes quantités de données sont produites à des taux toujours plus importants. Le séquençage d'un exome peut être terminé en quelques jours en utilisant le NGS, permettant la découverte de nouveaux variants en quelques semaines. La technologie génère un nombre considérable de faux positifs, et la différenciation des erreurs de séquençage des véritables mutations n'est pas une tâche simple. En outre, l'identification des changements d'intérêt parmi des dizaines de milliers de variants nécessite des annotations obtenues de nombreuses sources, ainsi que des capacités de filtrage avancées. Nous hébergeons la plate-forme technique de Génétique de la Faculté de Médecine de l'Université catholique de Louvain, mettant ces technologies de séquençage de l'ADN dernier cri à la disposition de nos scientifiques. Nous participons au développement régional et national des pipelines d'analyse. Nous fournissons également un soutien bioinformatique, essentiel pour l'analyse des données NGS. En outre, nous avons développé Highlander, un logiciel qui intègre plusieurs programmes et utilitaires in-silico d'analyse et disposant d'une interface graphique conviviale. Ceci améliore notre capacité d'identification et d'exploration des bases génétiques et épigénétiques des maladies. Nous développons également un LIMS (Laboratory Information Management System) qui nous permettra de gérer efficacement les données de nos biobanques, reliant les échantillons, les patients, les informations cliniques, les expériences, les résultats et les publications. Comme la bioinformatique devient une composante essentielle de la recherche génétique, d'autres projets in-silico seront déployés dans le futur.
Le domaine de la génétique humaine est actuellement révolutionné par le séquençage à haut débit ("Next Generation Sequencing" ou "NGS"). D'énormes quantités de données sont produites à des taux toujours plus importants. Le séquençage d'un exome peut être terminé en quelques jours en utilisant le NGS, permettant la découverte de nouveaux variants en quelques semaines. La technologie génère un nombre considérable de faux positifs, et la différenciation des erreurs de séquençage des véritables mutations n'est pas une tâche simple. En outre, l'identification des changements d'intérêt parmi des dizaines de milliers de variants nécessite des annotations obtenues de nombreuses sources, ainsi que des capacités de filtrage avancées. Nous hébergeons la plate-forme technique de Génétique de la Faculté de Médecine de l'Université catholique de Louvain, mettant ces technologies de séquençage de l'ADN dernier cri à la disposition de nos scientifiques. Nous participons au développement régional et national des pipelines d'analyse. Nous fournissons également un soutien bioinformatique, essentiel pour l'analyse des données NGS. En outre, nous avons développé Highlander, un logiciel qui intègre plusieurs programmes et utilitaires in-silico d'analyse et disposant d'une interface graphique conviviale. Ceci améliore notre capacité d'identification et d'exploration des bases génétiques et épigénétiques des maladies. Nous développons également un LIMS (Laboratory Information Management System) qui nous permettra de gérer efficacement les données de nos biobanques, reliant les échantillons, les patients, les informations cliniques, les expériences, les résultats et les publications. Comme la bioinformatique devient une composante essentielle de la recherche génétique, d'autres projets in-silico seront déployés dans le futur.
Le groupe de Génétique Moléculaire Humaine étudie les bases moléculaires et génétiques d'une variété de troubles du développement, y compris anomalies vasculaires, fentes labiales et/ou palatines, hypermobilité, ainsi que certains cancers, afin d'améliorer leur diagnostic et leur prise en charge clinique, et de découvrir de nouvelles cibles pour une thérapie. Notre laboratoire combine une grande bio-banque d'échantillons de sang et de tissus de patients, associée à de nombreuses données cliniques, des données de génotypage à grande échelle générées en utilisant des techniques de séquençage de pointe et un pipeline d'analyse des données développé en interne. La découverte de nouveaux gènes est suivie par la génération de modèles cellulaires et animaux, dans lesquels l'inactivation du gène coupable est utilisé pour comprendre les caractéristiques de la maladie chez l'humain. Ces modèles sont ensuite utilisés pour étudier les conséquences moléculaires et biologiques des changements génétiques responsables de la maladie, ainsi que pour tester des solutions thérapeutiques potentielles. Toutes nos recherches débutent avec des échantillons provenant de patients, et nous visons à faire le tour du cercle allant du lit au bench, et revenant au lit, en fournissant aux patients des améliorations fondées sur des connaissances viables et tangibles de leur santé.
 |
 |
 |
| Anomalies vasculaires | Lymphoedèmes | Fentes labiales et palatines |
|
|
|
| Cancer | Hypermobilité | Bioinformatique |
Nat Rev Dis Primers. 2021; 7(1):77.
Circ Res. 2021; 129(1):155-173.
Circ Res. 2021; 129(1):136-154.
JCI Insight. 2021; 6(15):e149831.
Hammer J, Seront E, Duez S, Dupont S, Van Damme A, Schmitz S, Hoyoux C, Chopinet C, Clapuyt P, Hammer F, Vikkula M, Boon LM.
Orphanet J Rare Dis. 2018; 13(1):191.
Quéré I, Nagot N, Vikkula M.
N Engl J Med. 2018; 378(21):2047-8.
Basha M, Demeer B, Revencu N, Helaers R, Theys S, Bou Saba S, Boute O, Devauchelle B, Francois G, Bayet B, Vikkula M.
J Med Genet. 2018; 55(7):449-58.
Amyere M, Revencu N, Helaers R, Pairet E, Baselga E, Cordisco M, Chung W, Dubois J, Lacour JP, Martorell L, Mazereeuw-Hautier J, Pyeritz RE, Amor DJ, Bisdorff A, Blei F, Bombei H, Dompmartin A, Brooks D, Dupont J, González-Enseñat MA, Frieden I, Gérard M, et al.
Circulation. 2017; 136(11):1037-48.
J Invest Dermatol. 2017; 137(1):207-16.
Am J Hum Genet. 2015; 97(6):914-21.
J Clin Invest. 2015; 125(9):3491-504.
Wassef M, Blei F, Adams D, Alomari A, Baselga E, Berenstein A, Burrows P, Frieden IJ, Garzon MC, Lopez-Gutierrez JC, Lord DJ, Mitchel S, Powell J, Prendiville J, Vikkula M; ISSVA Board and Scientific Committee.
Pediatrics. 2015; 136(1):e203-14.
Revencu N, Boon LM, Mendola A, Cordisco MR, Dubois J, Clapuyt P, Hammer F, Amor DJ, Irvine AD, Baselga E, Dompmartin A, Syed S, Martin-Santiago A, Ades L, Collins F, Smith J, Sandaradura S, Barrio VR, Burrows PE, Blei F, Cozzolino M, Brunetti-Pierri N, et al.
Hum Mutat. 2013; 34(12):1632-41.
Limaye N, Wouters V, Uebelhoer M, Tuominen M, Wirkkala R, Mulliken JB, Eklund L, Boon LM, Vikkula M.
Nat Genet. 2009; 41(1):118-24.
Jinnin M, Medici D, Park L, Limaye N, Liu Y, Boscolo E, Bischoff J, Vikkula M, Boye E, Olsen BR.
Nat Med. 2008; 14(11):1236-46.
Vikkula M, Boon LM, Carraway KL 3rd, Calvert JT, Diamonti AJ, Goumnerov B, Pasyk KA, Marchuk DA, Warman ML, Cantley LC, Mulliken JB, Olsen BR.
Cell. 1996; 87(7):1181-90.
Vikkula M, Mariman EC, Lui VC, Zhidkova NI, Tiller GE, Goldring MB, van Beersum SE, de Waal Malefijt MC, van den Hoogen FH, Ropers HH, et al.
Cell. 1995; 80(3):431-7.

 Telecharger
Telecharger
BASES GÉNÉTIQUES DES MALADIES HUMAINES
